多软件协同的Gromacs批量MD模拟计算
多软件协同的Gromacs批量MD模拟计算
此前我更新过几篇博客,是关于一些提高工作效率的小脚本,其实当时是想将它当作一个长期更新的帖子更新的,但是后面发现我写的小脚本越来越多,功能也愈发繁杂,涉及到的软件太多了,而且是shell语言加python语言杂乱着来的,功能更新也很快,就导致每次想写相关的博客的时候都感觉可能我可以把脚本写的更好再写,但是也就一直拖拖拖,到现在也没能继续完成,所以最近又开始了一些枯燥无味的批量计算任务,我决定不再拖沓,直接将写好的一些批处理脚本贴出来,并给出我自己开发的思路。
计算任务描述与总结
我个人觉得,当你需要用到批处理脚本的时候,你肯定对你需要进行的任务有相当深入的了解了,你能过将你要计算的任务拆分成合理的部分,然后熟练的对其进行每一步的处理,也就是说对于整个任务流程的每一步,你自己手动都能很轻松的完成,而批处理的计算任务之间的差异可能并不大,能过按照规律对任务进行调整。
计算任务
我这次需要计算的任务就挺简单的,计算的内容类似于我之前写的气体在孔径内的扩散,不过气体变成了单气体,然后需要调整的就是孔径,如果对气体吸附实验有一定了解的话,就可以知道这部分计算其实是为了验证分子探针测定孔径的实验结果。原理就是很简单的,我们通过不同的分子探针去测定分子在材料中的渗透效果,然后根据气体分子的动力学直径就可以得到一个参考的孔径,而计算的时候则可以用探针的气体分子来遍历孔径,然后得到一个理论上的孔径,这样就可以得到一个理论与实验相结合的结果。
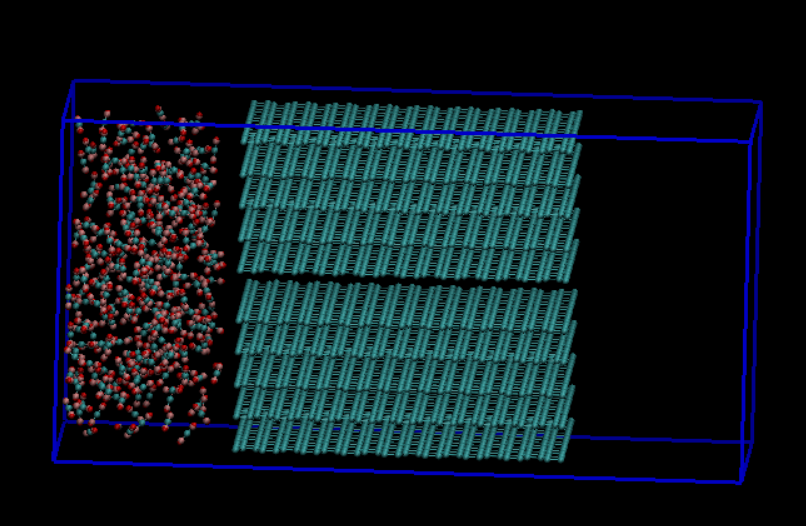
我这次进行的课题就是用N2和CO2作为探针,氮气不能过而二氧化碳能少量通过,所以需要用这两种气体分子去遍历计算不同的孔径,以获得碳层间的最佳距离,那么我们就可以用上次气体扩散类似的模型,左边的腔室添加适量的二氧化碳分子,然后调整中间炭材料的的孔径(层间距)大小来获取一个二氧化碳能过的最小孔径,然后以此结合实验来推定实验材料的孔径。
任务总结
根据上面的计算任务,接下来我们需要总结一下整个计算任务我们需要做什么,首先就是建模,这里我们需要三个模型,分别是N2、CO2和石墨碳的模型文件,这三个文件都很好获取,N2和CO2在trapEE small的拓扑文件里面直接就给出了对应的pdb格式的模型文件,而石墨碳模型则可以在MS里面自行建模一个,唯一的问题就是石墨碳中孔径的大小,这个其实是需要大量的计算来确定一个合理范围来进行探究,毕竟我们总不能从石墨层间距0.34一直增大到0.6nm这样大范围的孔径来筛选,计算太大,成本不能过接受,所幸我此前经过大量的计算,初步确定了氮气分子可能最小能过得孔径为0.56nm,所以我们可以以0.56nm为基点来创建模型,我这里建立了0.50nm-0.60nm范围内的孔径石墨模型,以0.01nm一个刻度,相当于一共创建了11个模型,这一部分虽然构建模型麻烦,但是的确找不到合适的批量处理方法。
将气体分子和石墨碳的模型创建好后,下一步就是将它们建立在一个盒子文件里面,这个就比较麻烦了,因为我们是通过packmol来生成盒子的,直接用packmol生成的pdb文件是不包括盒子信息的,而我们在计算的时候是需要一个真空腔的,而且如果炭材料以及气体分子离边界过近,会导致计算崩溃,所以此前我的模拟都是需要将packmol生成的pdb格式文件下载下来在vmd里面手动的设定盒子,然后再将所有原子向着盒子中央一定的距离才行,而现在我们每个气体按照孔径会有11个模型文件两种气体一共就会有22个模型文件需要处理,再用手动的去依次修改模型文件很明显有点浪费时间了,所以这里就要用到批量处理。
模型文件创建好了就是关于top文件的编写,这个也很简单,TrapEE small直接包含了N2和CO2气体分子的top文件,而石墨碳的top文件我在此前的博客里面也给出了,都是现成的,改一下分子数量基本就可以直接用,这里不多做赘述,而且top文件创建好了,对于所有模型的计算是可以共用的,所以操作量并不大,也不需要批量处理。
模型文件和拓扑文件都处理好了,接下来就是mdp文件,mdp文件很明显也是共用的,所以也无需批量处理,然后是提交任务,这部分很明显是需要对每个模型单独提交任务的,所以是需要批量处理的。计算完成后还有个数据的后处理,这个很明显也是需要批量处理的。
将上面的分析总结下来,大致上是有三个步骤需要我们进行批量处理的,分别是盒子模型的构建、批量提交任务以及批量后处理数据,这篇博客就仅做到前面任务提交的部分,由于后处理相对复杂,就以后再写了。
模拟计算任务全流程
根据前面的任务总结,我们需要对三个任务的步骤进行批量画处理,那么接下来我将写出我整个工作完成的步骤,其中也就囊括了不同软件的批量化处理,也会给出我之所以这样开发小脚本的思路。
分子模型的构建
分子模型的构建其实没有多少可以说的内容,两种气体分子的pdb文件是有现成的,石墨碳模型则是需要手动去构建模型,这里可以用MS的平移原子的功能来调整孔径,然后依次保存为不同孔径的pdb文件,最后我们是会获得13个模型文件(2个气体分子的模型+11个不同孔径的石墨碳模型),这部分操作没办法通过批量化操作,或者说批量化操作实现起来比较麻烦,花费大量时间去写这样一个脚本并不会节省太多的时间,因此可以手动建模。以下是我的工作目录下的模型文件的命名:

盒子模型的构建
盒子模型的构建分为两步,一步是通过packmol生成一个气体腔+石墨碳的盒子,第二步则是通过vmd构建指定盒子大小的,气体腔+石墨碳+真空腔的盒子模型,并且在这一步就将其转化为gro文件。这两步都需要我们进行批量处理。
使用相同配置的inp文件来批量运行packmol
我们可以直接用shell语言来调用packmol,但是批量运行的过程中要考虑的点主要是对模型文件的引用,比如我们用0.50nm孔径的石墨碳来构建盒子模型,这个就需要在packmol的配置文件里面指定该文件的文件名,那么当我们想继续使用相同的配置信息去为0.51nm的石墨碳生成盒子文件的时候,就需要将指定的文件名改为0.51nm的模型文件名,所以重点就是要遍历当前所有的石墨碳的文件,然后根据文件名来不断的修改packmol的输入文件,然后实现批量处理。
首先是packmol输入文件的编写,这里我的参数如下:
1 | |
我将输出文件定义为了box.pdb,然后对于给定的输入文件,这里是0.50nm的模型文件,将它固定在一个位置上,然后将300个气体分子(这里以二氧化碳为例)放在了石墨碳旁边的32*32*18尺寸大小的空间内,我将这个输入文件命名为mix.inp。
然后就是对批量处理脚本的编写,如下:
1 | |
这个批量处理的脚本最后写出来还是很简单的,我们之前对所有石墨碳模型的命名都是0**nm.pdb,所以可以用通配符*nm.pdb来轻松的定义当前目录下所有的石墨碳文件,其中*就表示任意长短的任意字符,然后对每一个文件,我们获得了它的文件名后将其传递给变量file,再用sed指定去定向的修改mix.inp文件里面的第四行的内容,而这一行内容就是packmol运行时指定石墨碳文件名的内容,这一行的内容改为了structure $file 的形式,其中$file表示引用这个变量,实际上它写入的时候会根据该变量的变化而变化。修改好mix.inp之后就是运行packmol,然后再根据我们石墨碳的模型文件名去设定一个输出文件名,这个规则就是在它的后面加一个_box,比如原本是0.50nm.pdb,那么我们输出的文件肯定不能再叫0.50nm.pdb了,就人为的设定为0.50nm_box.pdb,而之前我们其实在mix.inp文件里面指定了输出的文件名的,就是box.pdb,那么我们这里其实就是将每次输出的box.pdb更改为$file_box.pdb。写到这里我突然想到一个更加简单的脚本,那就是我们可以用修改输入文件的方法,直接修改packmol的输出文件名,而不是先输出一个box.pdb再改名的形式,代码如下:
1 | |
这样的代码可以更加简单。可以任选一份代码,将其保存为sh文件,比如我保存为batch_mix.sh文件。然后在工作目录下执行该文件:
1 | |
运行一会儿之后就会生成所有的box文件:
这样我们这一步工作就完成了,可以开始下一步的处理了。
使用tcl文件批量运行vmd来设定盒子并移动原子
vmd是支持用tcl文件来运行程序的,但是我对这个语言的了解并不多,不过它的逻辑还是比较简单易懂的,而且此前在vmd里面用的各种指令是可以直接用TK console执行的,所以真正的用起来还是没有想象中那么复杂。与之前的批量操作类似,首先肯定是遍历当前目录下所有的盒子模型文件,然后一个是要设定盒子的大小,二一个是要移动原子的位置,避免它们过于靠近盒子边界。
这个时候回顾一下之前用的指令,有了之前的计算经验,我这里就将盒子统一设置为{36 33 72}的大小,然后选中原子,再所有原子移动{1 0.5 1},这个都是我此前的计算得到的一个经验值,这也是为啥我说要对整个计算的所有步骤比较熟悉了才能实现批量化操作,这种数据只有经过比较丰富的计算才能获取到一个合理的值。
设定盒子的指令为:
1 | |
选中所有原子:
1 | |
将所有原子按照{1 0.5 1}移动:
1 | |
以上的操作就是我们要对所有文件实现的操作,其实第二步和第三步是可以合并的,我此前也是这样操作的,如下:
1 | |
这里我将它拆分开来了,用all这个变量来表示选中的所有原子,再将其传递给moveby指令,这是有原因的,因为我们后续还需要将所有结构保存为gro格式的文件,所有还需要选中所有原子,因此我们可以一开始就将其保存在all这个变量里面,方便我们后面输出数据。然后是读取文件,此前我们读取文件是用的以下的指令:
1 | |
这是代表我们直接新开一个vmd程序来打开055nm_box.pdb文件,还有一个方法使用以下的指令直接在当前的vmd进程里面打开模型文件:
1 | |
由于我们是要处理多个文件,所以我们需要在处理一个文件完成之后,将当前的模型文件关闭掉,就可以用以下的指令来关闭模型文件:
1 | |
这其中涉及到$mol_id,也就是我们打开的模型文件的编号,那么如何获取这个编号呢?我们可以在打开的时候就将其传递给变量mol_id:
1 | |
所以我们打开文件的时候就按照以上的方法来打开文件,也方便我们后续关闭该模型文件。然后是将模型文件输出为gro文件的格式,在输出之前我们还要定义输出文件的文件名,这部分的代码如下:
1 | |
这样对每个模型文件的处理都有了,接下来是遍历所有盒子文件的代码了,如下:
1 | |
我这里也是用*_box.pdb来表示所有的盒子模型文件,遍历所有文件,将每个文件名依次传递给变量file,所以最后的tcl文件就可以写成如下的内容:
1 | |
我将其保存为了all_rewrite.tcl文件,然后将这个文件保存到盒子模型文件所在的目录下,也就是工作目录下,然后就是在vmd里面去加载这个文件了,打开vmd,然后Extensions —> TK Console,打开对话框,
打开对话框后用cd切换到工作目录下,然后选中File —> Load file:
然后选中刚刚保存好all_rewrite.tcl,这样vmd就会自动执行这个文件里面的指令,很快就能获得生成的gro格式文件:
这一步后面最好抽样打开一些模型文件检查一下,确保没有啥问题了再开始下一步。

批量提交任务
经过前面的操作,我们可以很轻松的就获得我们计算所需的所有模型文件,而且直接是gro格式的,无需再做进一步的转化,接下来就可以开始计算了,计算之前我们还需要准备参数文件(mdp)以及拓扑文件(top),而且提交任务很明显也是要批量提交的。
输入文件的准备
mdp文件
mdp文件可以参考我的之前的博客内容,就是气体在炭材料内的扩散那一篇,mdp文件完全一致。
top文件
拓扑文件也可以参考一下我之前的博客,不过之前没有给出CO2的拓扑文件,所以这里贴一下,nonbonded的信息如下,TrapEE small给二氧化碳添加了两个虚拟点,所以这里的[ atomtypes ]有三个原子类型:
1 | |
将以上三个原子类型复制粘贴到原来的nonbonded.itp文件即可,然后是CO2.itp文件,如下:
1 | |
最后记得修改一下topol.top文件即可。
输入文件的验证以及获得整合后的top文件
对于所有文件,我们肯定要验证一下能不能正常生成tpr文件,我们可以任意挑选一个模型文件,将它单独放到一个目录下(改名为conf.gro),然后将拓扑文件复制过来,以及选择任意一个mdp文件,这里需要注意的是,由于计算的过程中我们是固定了某个组的,所以要确定固定的组所包含的原子正好是你想要固定的原子,这个可以用gmx make_ndx提前验证一下,执行以下指令:
1 | |
我返回的页面如下:
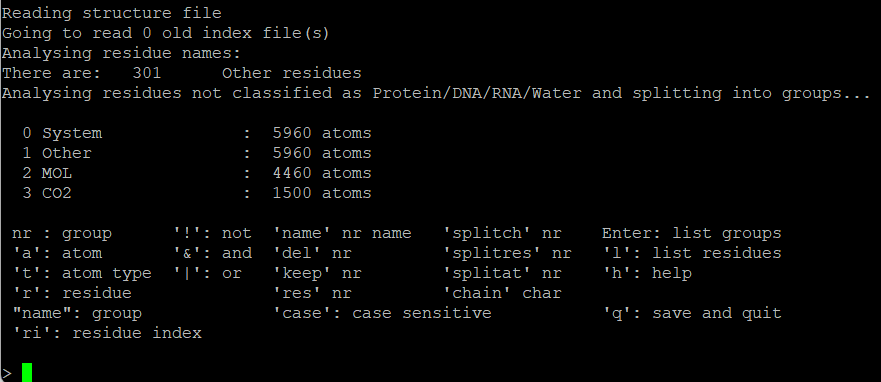
可以看到,MOL那个组别正好囊括了我们要固定的碳原子,而它是gro文件自带的分组,所以对于index.ndx文件并不是很需要,不过这里也可以直接输入q,也会得到一个index.ndx文件,后面引用这个文件也无伤大雅。
然后就是验证能不能正常生成tpr文件了,这个过程中我们正好可以直接将top文件整合了,方便后续其它计算直接使用,执行以下指令:
1 | |
由于我们之前将盒子模型的文件名改名为conf.gro,所以这里无需再指定结构文件,然后我们的top文件也是用的topol.top文件名,所以拓扑文件也无需指定,-f指定了输入的参数文件。-o指定输出文件名,-pp则是将整合好的top文件输出为min.top,-n则是引用index.ndx,这样的指令运行之后如下:
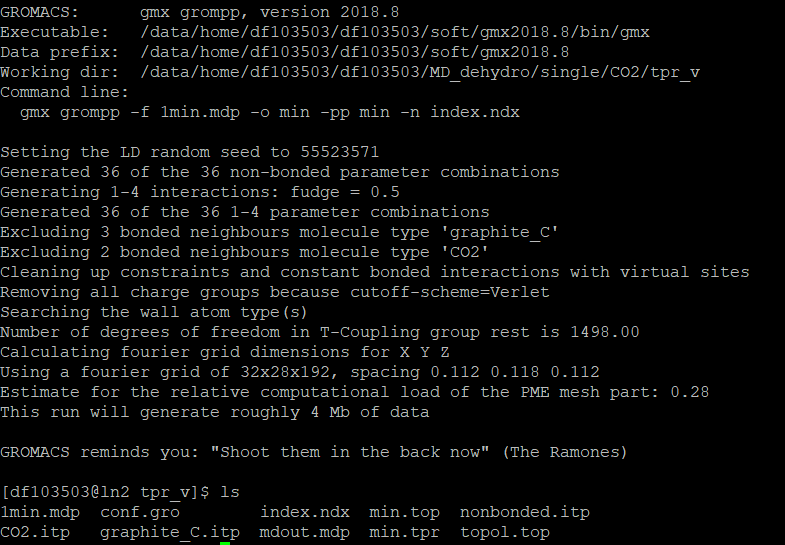
这就代表输入文件没啥问题,我们可以进行批量处理了。
批量提交任务进行计算
MD模拟一般都是分好几步运行的,所以我此前就已经写过将这好几个步骤整合起来,让它在运行的时候能过在一个步骤完成之后自动开始下一个步骤,而现在我们要在这个的基础上继续批量处理多个文件,所以就需要在外面再套一层,这里我们可以先将问题分开来看,首先是以前的脚本,我将其保存为gro_tot.sh,如下:
1 | |
这个脚本我就不多做赘述了,还是很简单的一个脚本,此前我的博客里面也有所讲解,可以根据自己的服务器情况将其修改,我在使用这个脚本运行任务的时候用的是如下的指令:
1 | |
现在我需要批量提交任务,就是根据不同的模型文件来分别创建文件夹,再将模型文件,参数文件,拓扑文件以及索引文件全部复制进去,然后再在各自的目录下用上面的指令来提交任务,首先遍历文件,这个在shell语言里面很简单:
1 | |
这样就是遍历所有以box.gro结尾的文件,然后将文件名传递给遍历i,然后是按照这个文件名创建文件并将各种文件复制进去,如下:
1 | |
dir_name就是将i这个变量的文件名删去它的_box.gro的后缀,然后创建文件夹,其中mdp、index文件以及top文件都是要重复使用的,所有用的cp,而结构文件是不重复使用的,所有我直接用的mv指令来移动,并且将其改名为conf.gro。
复制好之后就是切换到对应的目录,提交任务之后再返回:
1 | |
这样整合一下,批量提交任务的脚本如下,命名为batch_run.sh:
1 | |
这个脚本理论上来说就基本够用了,但是实际上使用的时候有一些痛点,首先就是这样做我们提交任务的脚本由原来的一个(gro_tot.sh)变成了现在的两个(gro_tot.sh+batch_run.sh),此外对于这样大批量的任务提交,我们在任务管理界面看不到不同孔径的任务信息,因为gro_tot.sh里面的#SBATCH -J Gromacs这一行的内容固定了,所有任务的任务名都为Gromac:
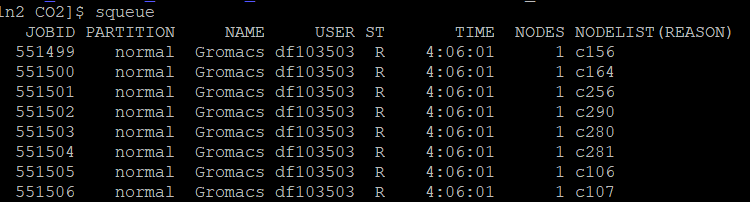
因此我们可以将其整合为一个文件,一来减少了文件的数量,二来可以将文件名这个变量传递给任务管理系统,让我们能够更好的检测任务的运行情况。这个整合也非常简单,可以直接用EOF的指令来囊括整个gro_tot.sh的内容,将其传递给sbatch,其中在#SBATCH -J Gromacs则可以将$dir_name这个变量替换Gromacs来实现任务名称的改变,修改后的完整代码如下:
1 | |
将其保存为batch_run.sh文件,然后复制到服务器里模型文件的存放目录下,再将之前准备好的mdp文件、整合好的min.top文件以及index.ndx文件也复制到这个目录下,接下来就可以执行以下的指令:
1 | |
注意,这里并不是用sbatch指令来提交任务,因为sbatch指令我们已经集成到batch_run.sh这个文件里面了,包括此前用两个文件分开的时候也是用bash或者sh来执行batch_run.sh文件即可。运行后的队列如下,可以看到任务名就能跟模型名对应上:
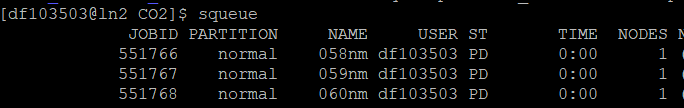
这样我们就实现了任务的批量提交了,接下来就是等计算完成再进行后处理了。
总结
以上就是到数据提交的全部批量处理了,基本上实现了任务的批量处理,对于系统单一变量的考察来说这样的批量处理已经足够了,我们可以很轻松快速的就对这样数十个模型完成模型的构建以及任务的提交,这样的思路也可以扩展到更加广泛的研究上,而且后处理也可以用类似的思路去完成,至少我们可以用数据去堆砌出一个像样的结果。