Gromacs2018的安装
Gromacs2018的安装
最近换到了外面的超算进行课题的计算,这个超算上自带的Gromacs并不好用,貌似都没有编译mpi,所以对于正常使用非常的不方便,所以我决定自己尝试着安装一遍
主要的安装步骤还是参考的sobereva的安装步骤
GROMACS的安装方法(含全程视频演示) - 思想家公社的门口:量子化学·分子模拟·二次元 (sobereva.com)
Cmake的安装
对于Cmake的安装我参考的是这个知乎的帖子:
直接在官网上下载了一个编译好的压缩包,上传到服务器上解压,然后配置了一下环境变量,主要原因是这个超算的cmake版本是2.x的版本,而Gromacs2018需要Cmake3.x的版本才能编译(突然明白了为啥这个超算没有对Gromacs进行mpi编译了)
直接用编译好的版本就非常方便了,到官网上下载对应的版本:
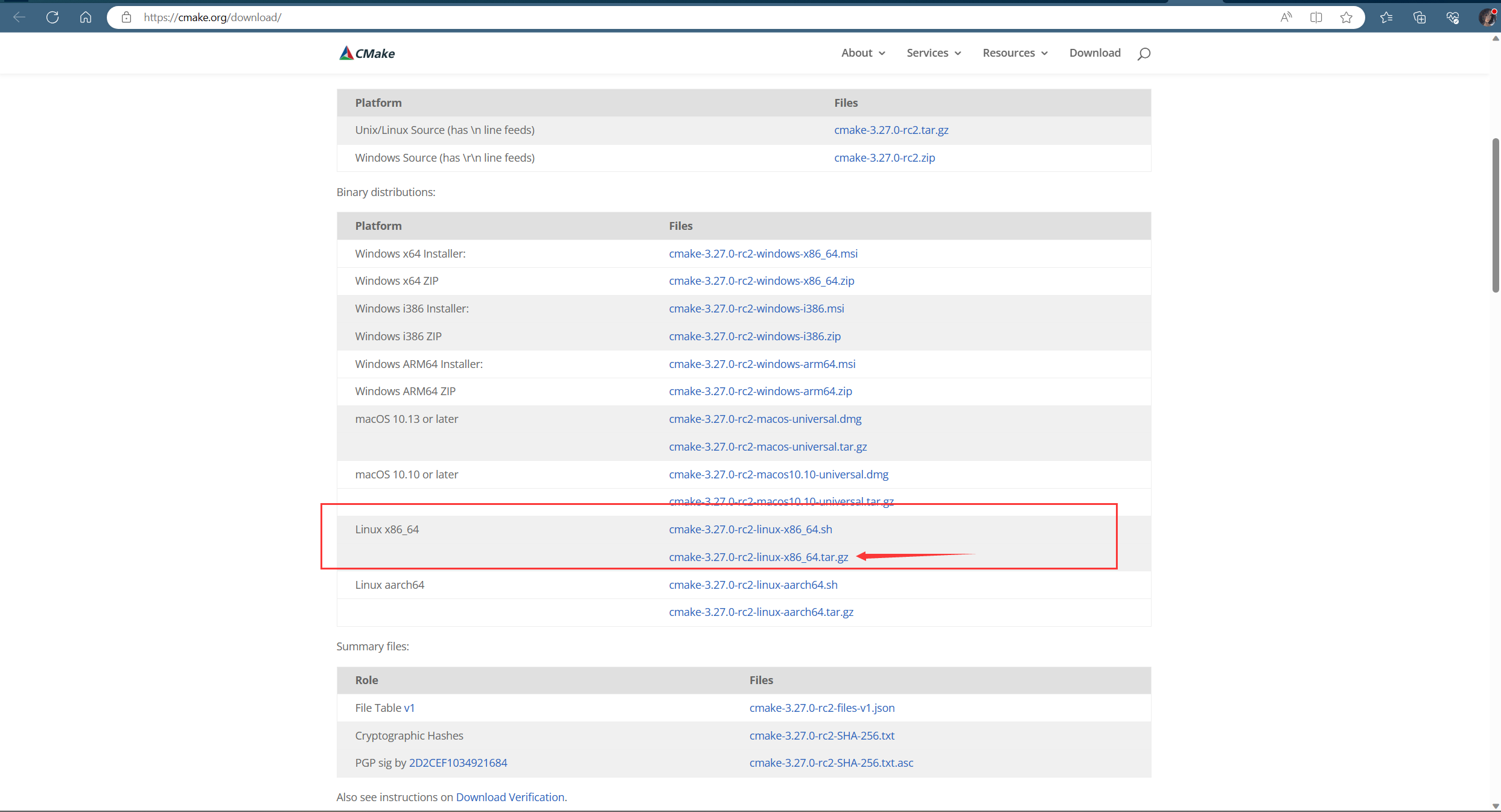
官网的下载页面链接在这儿:
下载好之后上传到服务器,解压:
1 | |
然后切换到解压后的目录下,用pwd查看当前路径:
1 | |
复制一下当前的路径,然后修改环境变量,将各个获得的路径添加到环境变量中:
1 | |
在.bashrc文件中添加:
1 | |
其中/data/home/df103503/df103503/soft/cmake-3.27.0-rc2-linux-x86_64/bin这个路径就是pwd返回的路径,这样修改环境变量就修改好了,然后source一下即可:
1 | |
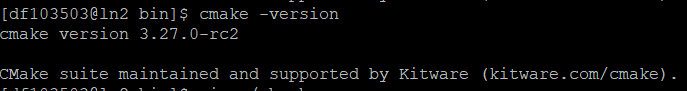
这样就配置好了,输入以下代码可以看看,如果返回的是3.27的版本,跟你下载的版本一致,那就配置好了:
1 | |
以下是我返回的值:
安装FFTW库
FFTW库在sobereva的帖子里面直接贴出来了,可以直接下载,不过这个也有官网的存在,可以去官网下载,但是sobereva的帖子中所说的是Gromacs2018要求的是FFTW 3.3.8,而官网上的版本为3.3.10:
我这里为了避免出错还是用的3.3.8,将这个压缩包上传到服务器,然后解压,解压之后进入到目录下,输入:
1 | |
其中~/soft/fftw338就是安装路径,我在最后将–enable-avx2加上了,我觉得这个服务器应该是支持AVX2指令集的。
这个执行:
1 | |
这个编译完成后就会多出来一个fftw338文件。
安装Gromacs
前面的准备步骤做完了就可以开始编译安装Gromacs了,将Gromacs的压缩包解压,然后进入到目录下,创建一个build的目录,依次的指令为:
1 | |
这样提前用cmake配置好之后,就可以用make进行编译了,继续执行指令:
1 | |
这个时候等一会就行了,很快就能编译完成。
关于指令集的修改
在按照上面的配置编译之后,后续在运行的过程中很可能服务器会提醒一个关于指令集的Note,这个可能会导致计算速度非常的慢,比如我这里在安装好之后,计算的过程中就有提醒:
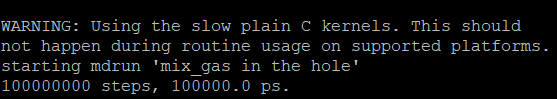
这就是指令集的问题,同时也会有这个提醒:
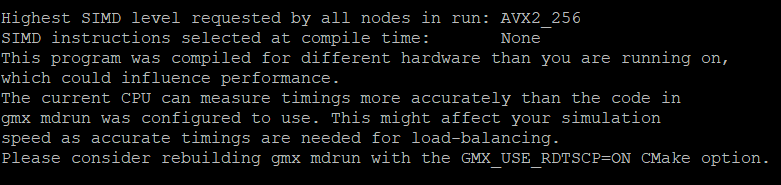
所以在cmake操作那一步还是需要有一定的调整,将原来的cmake指令改为如下的指令:
1 | |
在后面添加了-DGMX_SIMD=AVX2_256 和-DGMX_USE_RDTSCP=ON,关于-DGMX_USE_RDTSCP=ON由于是Gromacs软件建议添加的,具体添加了会引发什么效果也不清楚,但是这样添加之后对编译并没有什么影响。能够正常完成编译。
配置环境变量
接下来就是配置环境变量了,这个时候要注意,很可能你之前用过其它版本的Gromacs,已经加载过一些环境变量了,而这个时候加载的可能是临时的,所以会让你在这次登录的时候能够正常使用,而到下一次登录服务器的时候就提醒你缺少一些共享库了,所以还是要好好配置一下环境变量。
Gromacs需要的主要有两个环境,一个是gcc,一个是inteloneapi(这里我是参考的之前的版本配置好的文件内容),这个可以看看之前能正常运行的版本,一半运行之前会source一个文件,可以打开这个source的文件看看,比如我这里打开source的gmxbarc文件,出现了以下内容:
1 | |
最后一个就是我们配置gmx的环境变量,而前面两个则是配置gromacs运行所需的其它环境,因此理论上来说,我们需要将这两个环境变量也添加到我们自己的登录用户上,或者直接点,我们可以在自己的Gromacs文件中也写一个这样的gmxbarc文件,但是呢修改以下最后gromacs的路径。
首先,我们切换到刚刚我们编译好的Gromacs的目录下,进入到bin目录,然后pwd一下,复制返回的链接,然后直接在当前目录vim gmxbarc,创建一个gmxbarc文件,然后将上面的内容复制进去,最后的路径则改为刚刚复制的路径。操作如下:
1 | |
新的gmxbarc文件内容如下:
1 | |
这个时候gmxbarc的这个文件所在的路径就是我们刚刚pwd返回的路径,所以我们可以继续复制这个路径,然后将这个gmxbarc文件添加到环境变量中:
1 | |
在.bashrc的最后添加:
1 | |
最后source一下这个dot文件:
1 | |
这个时候环境变量就配置好了,gmx就可以正常使用了。