Gromacs计算气体在碳层间的扩散
Gromacs计算气体在碳层间的扩散
气体的筛分中,有一种方法就是通过炭材料来实现气体的高效筛分,而这个过程涉及到的是气体与炭材料之间的界面作用,这个过程很难通过DFT方法来对齐进行解释分析,所以可以通过MD方法来研究,所以通过Gromacs来研究这种固气界面的研究就很重要了,而且这样类似的研究也可以应用在电解液-电极材料的界面反应中。
计算的思路
对于气体在炭材料中的扩散,可以通过一些特定的方法简化,比如我们这里就是通过石墨结构来简单描述碳化之后的炭材料,然后通过调整石墨的层间距来实现不同大小的孔径:
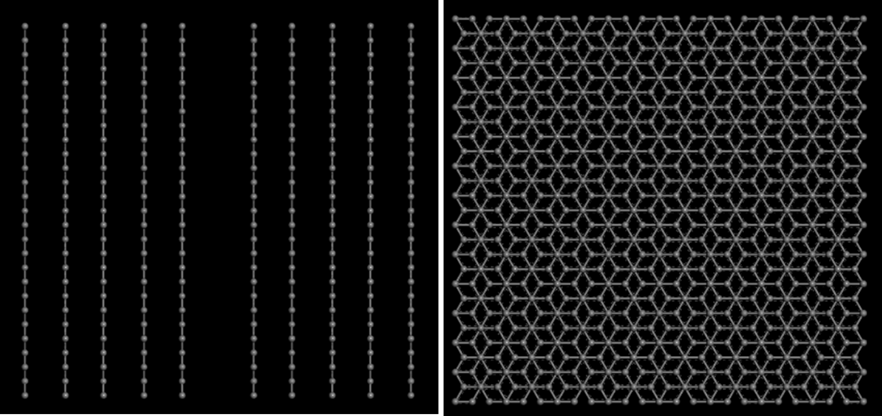
可以看到,这里用左右各5层石墨结构来表示炭材料的bulk区域,而中间的碳层间距则被我人为拉长了,这就是根据实验的结果来构建的孔,这个数据一般跟实验上氮吸附结果是无法对应的,氮吸附等气体吸附方法测量出来的孔径是没有考虑范德华力的校正的,所以一般来说还需要对一个碳层增加0.17nm的范德华力半径校正,左右两层则需要添加0.34nm的校正。
注意,这里可以看到我构建的模型孔洞方向是沿着z轴方向的,这样构建的原因是gromacs的MD模拟中,对于非周期性的模拟,进支持z轴方向的非周期性,而我们的气体扩散模拟,对于孔洞方向是扩散的方向,是不需要周期性的,所以这里就需要将孔洞方向与z轴一致,才能实现气体的筛分模拟,为了便于理解,我绘制了一个草图:
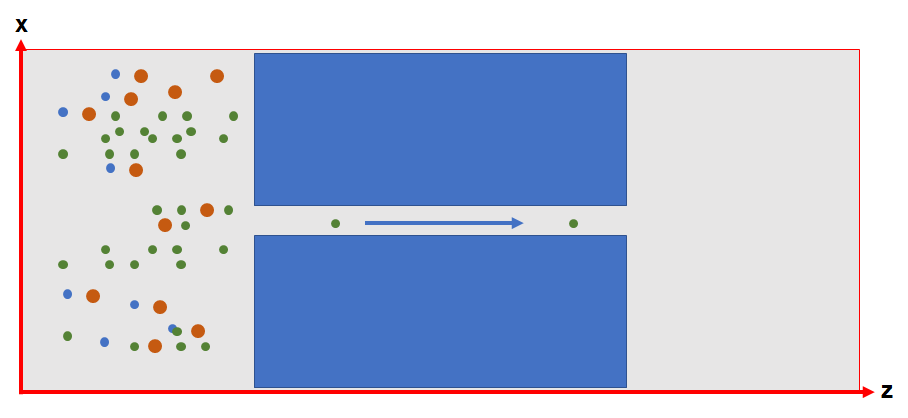
这样看起来就比较直观了,最外面的红色框就是代表整个模拟的box,内部蓝色的区块就是炭材料,区块中间的空白就代表炭材料的孔洞,box左侧的小球就代表气体分子,气体分子则向着箭头方向扩散,到达右侧的空腔,这里就能更好的解释为什么要z轴的非周期性,如果z轴方向也是有周期性的话,那么它的左侧就可以很容易的直接就自由扩散,经过盒子边界到达右侧的空腔,自然也就不存在什么气体筛分的过程了,所以z轴这里就需要一个非周期性,让气体扩散到右侧空腔的过程,必须要经过中间的碳层。
Gromacs实现xy周期以及固定碳层
Gromacs实现xy周期性的办法,或者说大部分软件实现xy周期性的方法都比较简单粗暴,那就是在z轴的两端,或者一端,加上一堵墙即可,这堵墙阻隔了边界内的原子向边界外的移动,就可以防止原子移动到另外一段,从而移除了这个方向的周期性。Gromacs的可以通过在mdp文件里面加一些与wall相关的关键词来实现这样的功能,这里贴一下我实现wall的代码:
1 | |
pbc设置的就是周期性,一般这个关键词默认是xyz,我们这里需要将其修改为xy,而有关wall的各项参数设定,可以参考这篇文章:
GMX中墙与冻结组的设置 - 简书 (jianshu.com)
当然也可以参考gromacs的手册;我这里为了保险起见,设定了两个wall,也就是左右各有一个,然后采用的直接的L-J势函数描述墙与气体分子的相互作用,这里wall-atomtype我设定的是碳原子的原子类型,也就是我们模拟炭材料时所用的原子类型。
接下来说说关于炭材料的固定,我们这里的模拟由于仅需要考虑的是炭材料对气体的筛分作用,或者准确的来说是炭材料孔径对气体的筛分作用。所以炭材料内部的结构变化我们是不需要考虑的,而且这也是为啥我们在一开始建模的时候并没有对炭材料的边缘进行加氢饱和处理的原因。所以对于整个炭材料来说,我们模拟的过程中不需要它移动,只需要它固定在初始位置上提供与气体分子的相互作用力就足够了,而gromacs中是提供了这样的固定原子的关键词的,如下:
1 | |
这个关键词很好理解,第一个freezegrps即代表要固定的组是啥,这个是自定义的名称,需要我们在写好mdp文件的时候为炭材料创建一个名为MOL的组,将所有需要固定的碳原子包括在这个组内,第二个freezedim则代表固定的维度,对于完全不需要移动的碳原子,那么三个维度都要固定,自然也就是三个Y。
Gromacs实现模拟的全过程
为了更好的完成整个教程,加上近期正好需要再进一步计算,所以这里就对这个全过程进行一个记录,方便看我博客的人能够系统的了解一下整个工作流程,这里的整个工作流程是基于我自己平时的工作流确定的,有些步骤不是必须的,有些步骤也是可以通过其它方法实现的,所以可以酌情学习,如有遗漏的地方,敬请海涵。
模型文件的生成
模型文件的搭建分为三个部分,第一个是炭材料的构建,第二个是气体分子模型的构建,第三个则是整体盒子的搭建,这里就涉及到多个软件的协同作用了。
1. 炭材料模型的搭建
炭材料模型的搭建其实比较简单的,大体上就是我在前面贴出来的模型图,在MS中导入一个石墨的结构,然后通过矩阵变化转变为正交晶格,再扩胞,然后调整一下层间距即可,最后转化一下坐标轴,将孔洞方向调整为z轴,我这里就不多做演示了,因为写这个步骤感觉可以再写一篇博客了,很基础的内容,但是想讲完的话内容也挺多的。大致上是需要获得前面我贴出来的一个模型即可,注意,这里还有个细节就是我们需要通过MS里面测量距离的工具,确定整个炭材料的模型大小,就是它的xyz轴各长多少,这对于后面我们构建盒子来说很重要。
2. 气体分子的搭建
气体分子的搭建就相对复杂一些了,我这里模拟的是一个甲烷,氮气,氢气的混合气体,对于这些小分子气体,它们在模拟所用的结构文件跟拓扑文件关系很大,分为UA(Unit Atom)和AA(All Atom),而且对于一些AA拓扑文件来说,还有一些虚拟点需要定义,所以我们在打算模拟气体小分子的时候一定是先要获取到它的合理的拓扑文件,再进行模型构建。
这里就用TraPPE的氮气来举例,在TraPPE力场的网站上直接搜索氮气即可得到相关力场的参数信息,TraPPE力场的网站如下:
TraPPE: Transferable Potentials for Phase Equilibria Force Field (umn.edu)
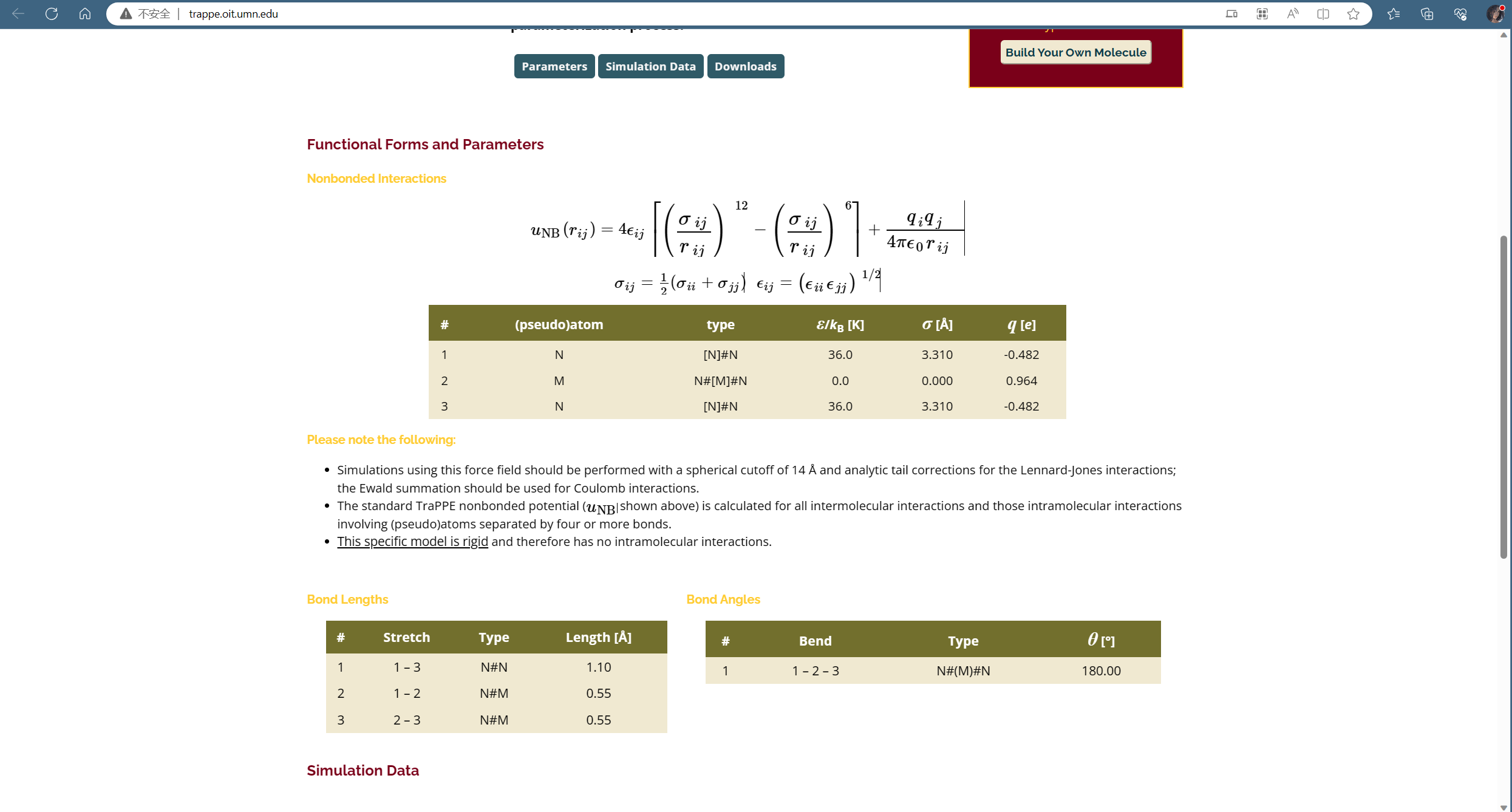
不过官网上有关结构的参数信息是表格形式的,是需要自己根据它的参数将其写成Gromacs的拓扑文件的,还是比较麻烦的,不过所幸我在一个论坛上找到了前人总结的TraPPE气体小分子力场文件的压缩包,里面就有TraPPE所包含的各种小分子(small)的拓扑文件,那篇帖子的链接如下:
TRAPPE_small力场包 - 分子模拟 (Molecular Modeling) - 计算化学公社 (keinsci.com)
在这个压缩包里面就有各类小分子的拓扑文件,找到氮气的itp文件,文件内容如下:
1 | |
安装包中同时也给出了氮气分子的结构文件(pdb格式),所以可以直接使用:
1 | |
不过这里我们也可以自己建立一个氮气的分子结构。氮气的模拟是All Atom的,而且有一个虚拟点,所以建模的时候我们需要将这个虚拟点位建模出来,在TraPPE的网站上有对氮气分子的结构参数的描述,就是一个直线型的分子结构,虚拟点在两个氮原子的中央,那么我们就可以通过MS画一个这样的结构,中间的虚拟点可以先用随便一个原子代替,然后修改原子信息来认定它为虚拟点,先在MS里面绘制一个如下图的结构,可以通过MS的角度调整和键长调整功能来设定指定的键角和键长:
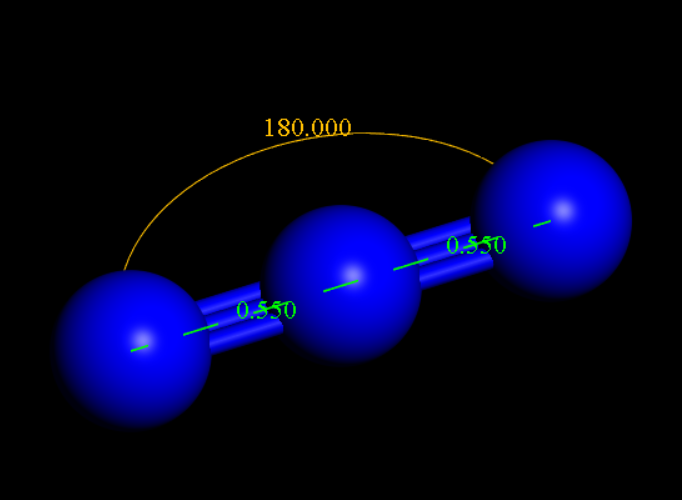
然后选中中间的原子,在左边的properties中找到Composition,双击它,在弹出的设置框中,将Name改为MW,如下图:
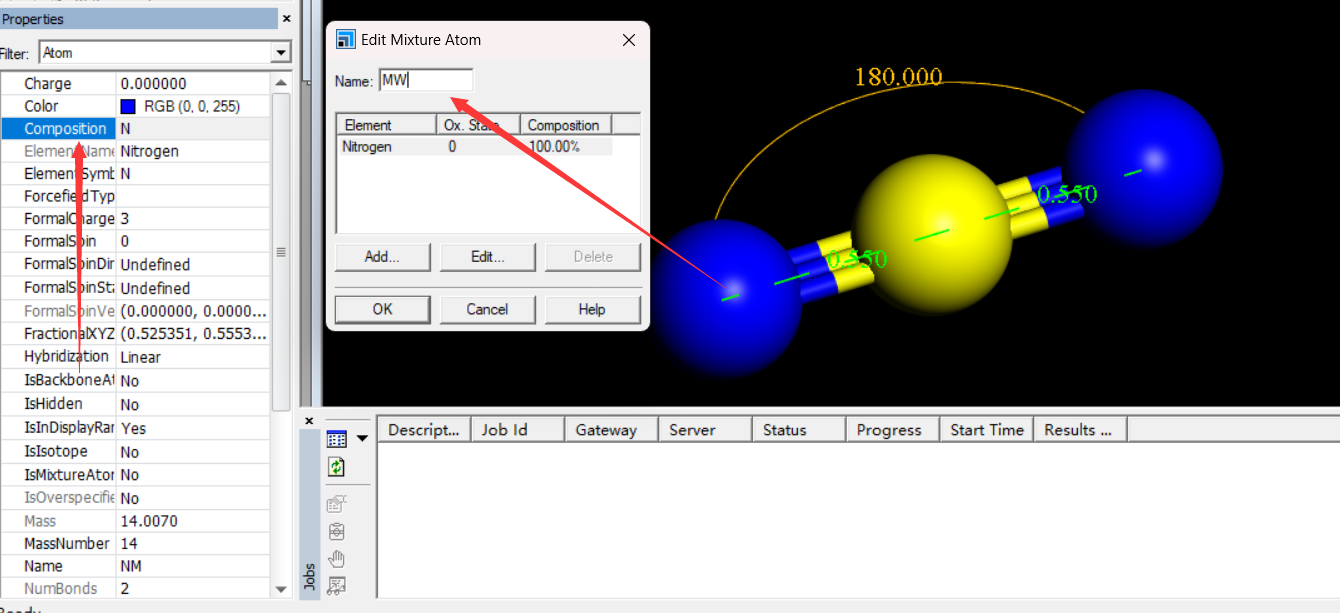
然后点击OK,即可成功设定,然后导出pdb格式的文件,这个时候可以用文本编辑器打开pdb格式的文件,对它进行一些修改,主要是为了删去一些多余的信息并且调整原子的排序,刚开始导出的pdb文件内容如下:
1 | |
将它的原子排序以及分类名称改成与拓扑文件相对应的,避免后续生成tpr文件的时候报错,然后删去多余的信息,我这里修改之后的文件内容如下:
1 | |
这样我们就获得了氮气的分子模型,此外再说一下氢气和甲烷的分子模型,对于甲烷气体来说,一个用的比较多的是opls-aa的力场,这个看名字也知道,是AA的类型,但是我在相关的文献中没有找到用这个力场的,主要是论坛中推荐的比较多,此外就是TraPPE网站上的甲烷力场,在TraPPE上有两种类型的甲烷力场,一个是常规的UA力场,即将整个甲烷分子看作一个粒子来进行模拟,还有一种则是EH(explicit hydrogen)力场,EH也就是所谓的显式氢力场在液相的描述上更有优势,但是应该会增加计算量,对于我们的计算量来说使用UA力场就足够了,所以可以直接用UA力场,这样的话我们就只需要指定一个原子来表示甲烷分子即可,可以直接copy前面的pdb文件,然后删去其它原子,仅保留一个原子,然后改一下原子的种类即可,我最后得到的甲烷的pdb文件如下:
1 | |
类似的,根据参考文献中,我们对氢气也是采用UA的力场,将氢气看作一个粒子,参考文献如下:
不过这篇文章中对氢气的L-J势函数也是引用的另外一篇文献,如下:
所以我们根据这样的UA力场,也可以用一个粒子来表示氢气分子,我最后的氢气分子的pdb文件如下:
1 | |
3. 整体盒子模型的搭建
根据前面的一系列操作,我们将会有四个文件,一个是炭材料的模型文件,然后是三个气体分子的模型文件,将这四个文件分别命名为hole.pdb, H2.itp, N2.itp, Ch4.itp。
我们可以将其上传到服务器上,通过packmol来生成盒子的pdb文件,使用packmol的时候需要一个.inp文件来设定生成的一些参数,这个时候就需要我们对自己需要生成的盒子大小有一个大概的想法了,根据前面的草图,我们需要将hole.itp文件放到整个盒子的正中央,然后将气体分子随机分布到文件的一侧,这里可以用packmol的一些特殊的关键词来将其固定生成到中间,以下是我用的mix.itp:
1 | |
对前面的参数我就不多做赘述,可以去看packmol的操作手册,这里需要注意的是各个模型的定位参数,尤其式hole.itp固定的坐标,它是以整个模型的中心点来定位该模型的,这个时候最好是画一个草图来确定该模型中心点在盒子中的坐标,而整个盒子又不能太大,因为在xy轴方向还是周期性的,如果盒子的xy轴过大,则会导致在材料与边界处的距离过大,气体就会从这个距离中通过,也就是盒子xy轴的长度减去hole模型xy轴的长度应该要小于孔的层间距离。随后就是气体的分布空间,这个空间要考虑到气体的分子数量以及整体盒子的大小,整个盒子不能太长,太长的画在模拟的时候会崩溃,而太短的话则可能会导致气体数量过少,气体的数量还需要根据实验的气压进行计算,所以这个步骤也是需要进行一定的提前计算的,我这里是提前按照1bar的气压进行了计算,得到了一个比较合理的气体分子数量和盒子腔体大小。
确定好.inp文件没有问题之后,我们将其保存好,命名为mix.inp,然后将所有模型文件和mix.inp文件放在同一目录下,输入:
1 | |
很快即可获得一个盒子模型,但是这个时候的模型还不能直接用,需要将其导出到vmd中进一步调整盒子大小,将输出的box.pdb文件传到电脑上,用打开vmd,在弹出的shell中切换到保存该box.pdb文件的目录下,然后:
1 | |
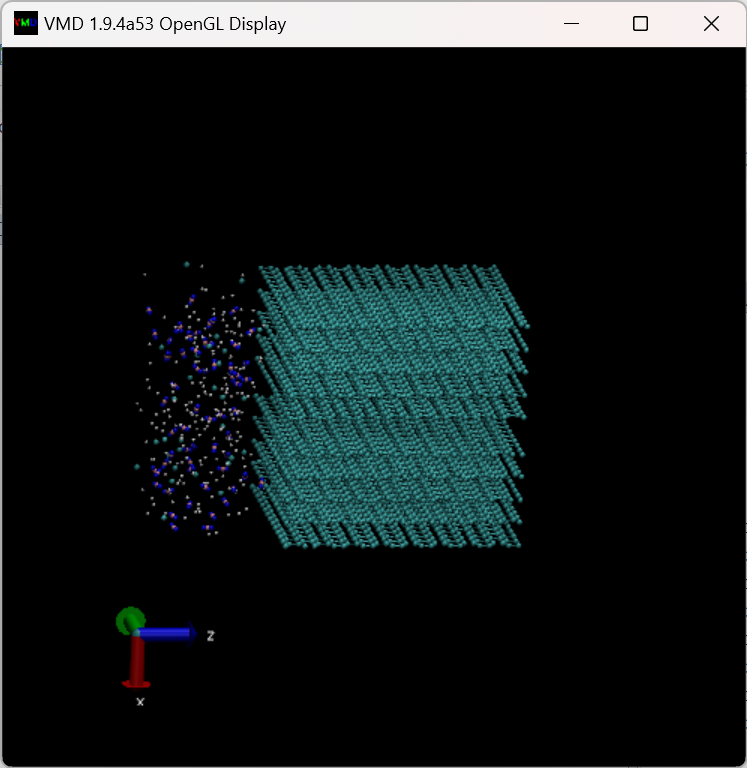
接下来先调整一下视角和原子的显示类型,在Display页面选中Orthographic,然后在Graphics中选中Representations,在弹出的对话框中找到Drawing Method,选中CPK,再点击Apply即可,这个时候模型就变成了如下所示:
接下来就是对其进行一系列的操作来设定盒子并且让它处于盒子中合理的位置,首先在vmd的shell中输入:
1 | |
这样即可以在模型中显示盒子,这个时候的盒子仅包括了气体分子和炭材料,并没有另外一段的空腔,所以我们要重新设定盒子的大小来设定空腔,根据之前的我们对体系的定位,将左边气体腔跟右边气体腔的大小一致,那么最后可以得到一个总的盒子大小,在shell中输入:
1 | |
这个时候可以在模型中看到,盒子扩大了,但是整个模型贴近一个角落,如下图:
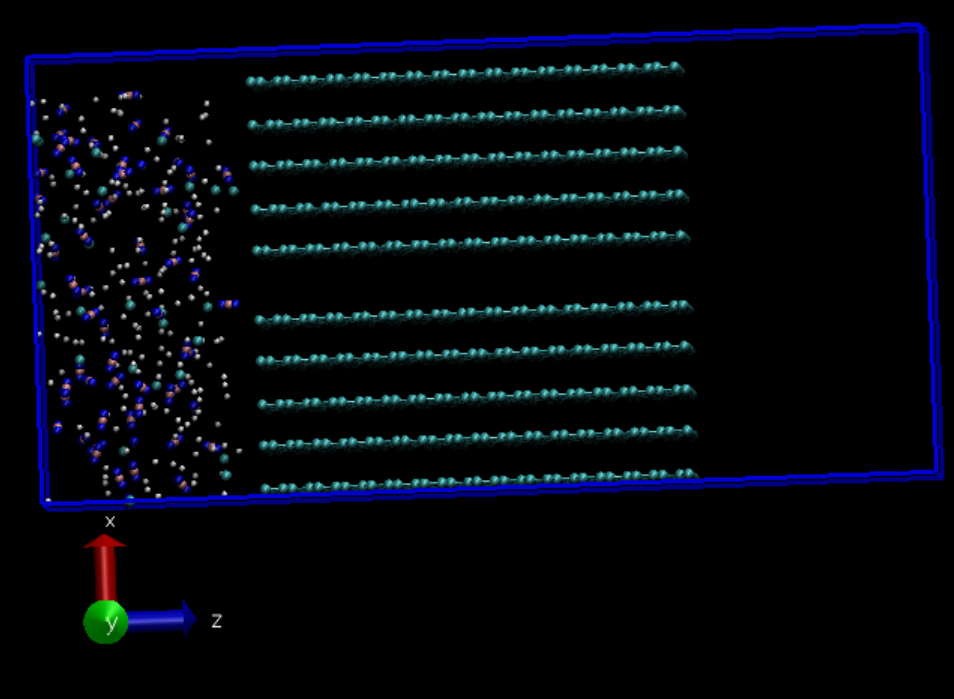
所以我们还需要将整体模型向中间移动一定的距离,可以通过指令来移动所有原子,输入:
1 | |
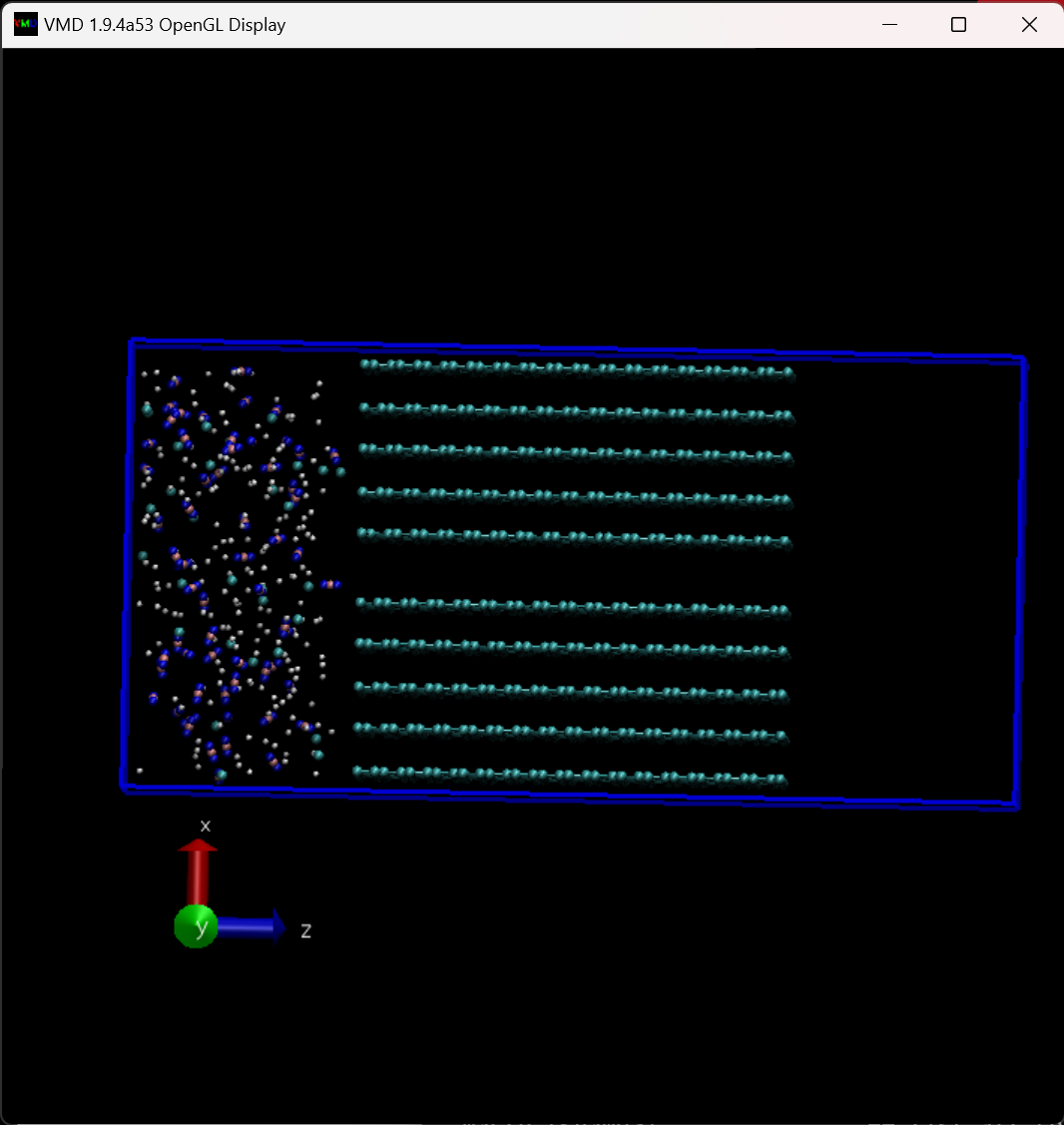
这样就是让所有原子在xyz三个轴方向均移动1埃的距离,这个时候整个模型的所有原子就会处在一个比较合理的位置了,这样的操作也是为了避免后面在模拟的时候气体分子离wall过近而导致计算崩溃。最后得到的模型如下:
最后就是在main的界面中选中文件,然后点击File,选中Save Coordinates:
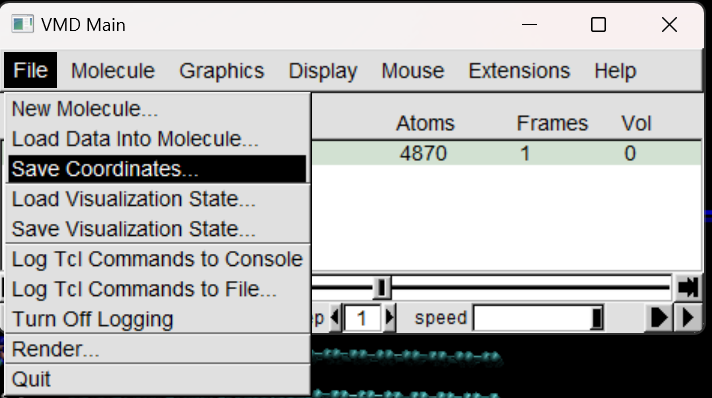
然后在在弹出的对话框中,在Selected atoms中选中all,在File Type中选择gro,最后点击Save即可成功将其转化为gro文件保存,这样我们也就能获得模拟所需的模型文件了。
拓扑文件的编写
拓扑文件与结构文件类似,就是有几个模型文件,那么我们就需要几个对应的拓扑文件来描述它们的相互作用,不过在构建模型的时候,其实我们对气体小分子的模型文件已经找得差不多了,所以这一步编写就好写了,而对于炭材料,由于它是一个非常大的体系,但是里面却全是炭原子,而且碳原子是固定的,我们不需要考虑碳原子之间的键长以及键角之类的信息,所以我们对它就可以将其看作是数个碳原子单分子的集合,那么在写这个炭材料的itp文件的时候就能简化许多。
1. 炭材料的itp文件
前面说了,我们将炭材料看作是一个单分子,那么我们就只需要指定一个碳原子的L-J势函数的参数信息即可,键长键角则可以完全不用考虑,如下:
1 | |
这里我将nonbonded.itp里面石墨碳的[ atomtypes ]复制了过来,正常写的话graphite_C.itp仅包括[ moleculetype ]以及[ atoms ],对于[ atomtypes ]里面的sigma和epsilon的值,我是参考的一篇文献:
Nanoparticle adhesion in proton exchange membrane fuel cell electrodes - ScienceDirect
这样我们在后续的过程中就可以直接使用这个单分子碳的拓扑文件,在后面指定分子数量的时候就看我么的炭材料模型有多少个碳原子即可。
2. 气体分子的itp文件
气体分子的itp文件在前面其实已经说得差不多了,具体的来源在前面建模已经说得很清楚了,我这里也是将nonbonded.itp里面的[ atomtypes ]复制了出来方便查看,各个分子的力场信息如下:
N2:
1 | |
CH4:
1 | |
H2:
1 | |
可以看到,对于UA来说,粒子的质量(mass)即是其分子的质量。
3. 最后topol.top文件以及nonbonded.itp文件
最后将所有文件都重新罗列一遍吧,方便读者取用。
nonbonded.itp:
1 | |
graphite_C.itp:
1 | |
N2.itp:
1 | |
CH4.itp:
1 | |
H2.itp:
1 | |
topol.top:
1 | |
计算参数mdp文件的编写
准备好结构文件和拓扑文件之后,我们就可以准备计算所需的参数文件了,这个涉及到我们该怎么算的问题,前面已经说了,我们需要通过Gromacs来实现对炭材料的固定,并且添加两个wall来去掉z轴方向的周期性。这里有个点就是我们在气体没有填充的那一端,开始模拟的时候是空腔,这也是左右两端存在一个压力差,也就给气体的扩散提供了一个推动力,但是这个空腔对于NPT系综来说是致命的,如果传质不及时的话会导致盒子的剧烈变形,而引发模拟的崩溃,所以对于这个体系显而易见的,我们只能采用NVT系综,而且对于这种带有氢气的分子,要特别小心氢气的活泼性,它太好动了,以至于能够在极短的步长内就跑到一个不合理的位置,从而导致模拟的崩溃,所以一开始的时候要充分的最小化,并且开始模拟的时候步长一定要控制在一个较小的值,充分预平衡之后再进行较大步长的成品模拟。
1. 能量最小化
能量最小化的mdp文件其实没有特别多需要注意的地方,主要是一个在我看来比较玄学的设定就是,先用steep方法能量最小化,然后再采用cg方法能量最小化,据说这样可以让能量更快的达到一个合理值。而对于库仑力以及范德华力计算半径的设定则注意,不能超过盒子最小边长的一半。以下是我能量最小化所用的mdp文件:
1 | |
对于第二步的能量最小化就是将第一步的integrator= steep改为integrator= cg即可。
2. 小步长下的预平衡以及成品模拟
对于预平衡,这里反复强调一定要用小步长,这是我血与泪的教训,之前反复的报错,各种找原因,最后就是出在了开始的步长这个问题上。预平衡跟成品计算的mdp文件是差不多的,我这里采用了Nose-Hoover热浴的方法,然后步长设定为0.01fs—> 0.1fs—> 1fs,最后的1fs就是成品模拟了,前面两个步长各跑500000步来让体系充分预平衡,最后再跑一亿(100000000)步,也就是100ns作为最终的成品模拟,来进行数据分析。以下是第一次预平衡的mdp文件:
1 | |
对于第一次预平衡涉及到了一个初始速度的生成,而对于预平衡,我们不是很需要去研究它的轨迹,而是对它的能量等信息比较看重(可以看是否充分预平衡),所以对于nstenergy和nstlog就可以设置得稍微小一些,而对于输出坐标信息间隔长度的参数nstxout-compressed就可以设置得大一些。以下是第二次预平衡的mdp文件:
1 | |
第二次预平衡是继承了上一次预平衡的gro文件继续计算,保留了速度信息,所以就不需要生成速度的参数了,而这一次预平衡为了提高效率,则可以在上一步的基础上提高步长来让体系充分预平衡。最后是成品模拟的mdp文件:
1 | |
在最后的成品模拟中,我们需要得到气体的扩散信息,因此需要记录较为详细的坐标信息,就可以将nstxout-compressed设置得大一些。
生成tpr文件并提交计算
在提交计算之前,我们需要根据前面已有得gro文件、拓扑文件和mdp文件先生成tpr文件,然后再运行gromacs进行计算,这里涉及到多步的计算,所以我们可以先生成tpr验证文件没有出错,然后再重新编写提交任务的脚本文件,让它能够在节点上连续的进行多步计算。
运行grompp指令验证文件的合理性
为了确保gromacs计算的过程中不会因为设置或者文件配置等问题出错,我们得先验证它能否生成tpr文件,将前面我们生成得box.gro文件改名为conf.gro,然后将所有的拓扑文件和任意一个mdp文件放到同一个目录,比如我这里就是将1min.mdp文件以及一系列的itp文件,然后一个名字为topol.top文件还有结构文件conf.gro文件存放在了同一个目录下:

这个时候需要注意的是,我们是否自带MOL这个组,我之所以将所有碳原子在一起的组命名为MOL,并不是随机命名的,是因为我通过MS建模之后,以及后续一系列的操作,自动会给C原子分到一个叫做MOL的组里面,如果你是通过其它建模软件或者中间有些其它操作的话,你的体系中就很可能没有这个组,或者说MOL组里面有其它的原子,这个时候可以用gmx的make_ndx看一下是否需要创建一个index.ndx文件,输入:
1 | |
我这里的gmx指令是通过gmx_mpi调用的,这是编译的时候的遗留问题,影响不大,跟版本也有一定的关系,有些服务器的gromacs则是可以直接通过gmx来调用。
然后会进入一个创建组的页面,我这里返回的信息如图:
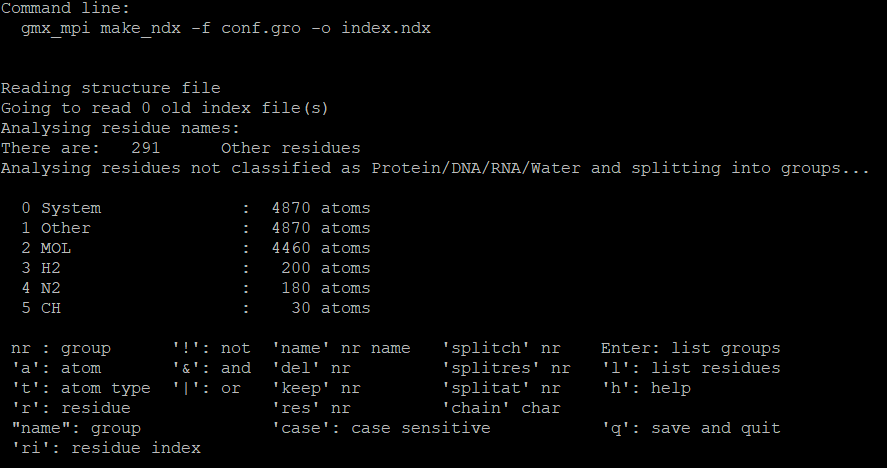
可以看到MOL组里面的原子数量正好是碳原子的数量,也就是说MOL这个组正好就将体系中的碳原子包括进去了,所以我就可以不用索引文件(index.ndx)来指定新的组,如果你这里没有任何一个组能够囊括所有碳原子而不会包含其它原子,那你就需要新建一个组,可以通过继续输入a C的指令来将左右碳原子分配一个组,并且这里的组的名称就变成了C,当然也有其它的指定组的方法,具体可以参考程序文档,创建好组之后输入q保存并退出,就会产生一个index.ndx的索引文件。
确定好分组的问题之后,就可以执行gmx grompp的指令了,指令如下:
1 | |
注意,这个是一个极简的指令,并没有指定结构文件和拓扑文件,这也是为啥我们在开始会将结构文件改为conf.gro,以及将拓扑文件保存为topol.top文件的原因,当你没有指定结构文件和拓扑文件的时候,gromacs会模型结构文件为conf.gro,默认拓扑文件为topol.top,而且我还省略了文件的后缀名,这样都是为了减少输入量,此外,如果你之前进行了分组的话,那么这里就需要在后面加上索引文件才能让gromacs得到分组的信息,那么指令就如下:
1 | |
我这里运行有一个Note,是关于原子冻结的,对计算没啥影响,可以忽略:
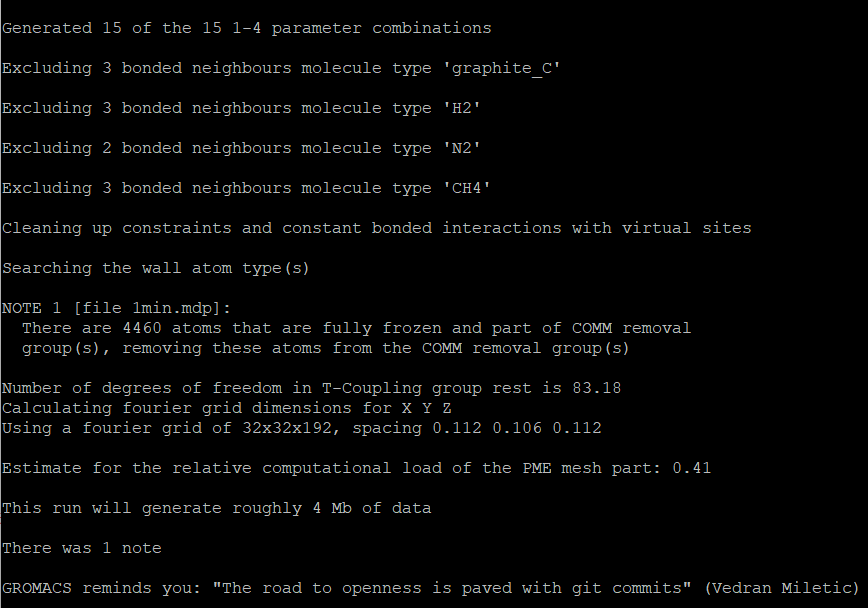
这样就代表文件生成没有什么问题了,目录下会多出三个文件,分别为1min.tpr,min.top以及mdout.mdp文件,分别是计算的输入文件,整合到一起的top文件和将所有参数罗列出来的mdp文件。对于min.top文件,我们生成过一次,后续的计算就都可以用这个top文件进行计算了。
编写任务脚本实现在节点上的连续计算
我用的服务器是slrum任务管理系统,通过sbatch指令提交一个sh文件来提交任务,所以我们可以修改sh文件来实现在节点上的连续计算,根据之前的讲解,我们将整个计算分成了5步,两次能量最小化,两次预平衡以及最后的成品模拟。
最后在模拟的时候出了一些小意外,为此我还自学了如何安装Gromacs2018,主要是现在这个新的服务器不支持mpi rank导致计算的时候会崩溃,所以我重新在自己的账号上编译了一个Gromcas2018,具体可以参考我下一篇博客,配置好之后调用gromacs的指令也就变成了gmx了,所以后续的脚本运行指令都是gmx了。
前面说了为了让成品模拟不崩溃,所以我们分五步计算,那么任务脚本的首要任务肯定是创建五个文件夹来分别存放五次计算任务的文件,我写的内容如下:
1 | |
前面四行的#SBATCH是有关提交任务的设置,需要根据服务器的需求以及节点等自行更改,创建好5个文件夹之后,我们就可以将现有的文件分配给各个文件夹,前面我们已经得到了整合之后的min.top文件作为整个计算的top文件了,那么我们这里所有步骤计算都可以用这一个拓扑文件,还有就是将各个步骤的mdp文件也分配给各个目录,此外对于第一步能量最小化,就可以将我们的初始结构conf.gro分配给它,然后将它计算得到的gro文件再依次复制给后续的文件完成计算,对于分配文件,则是以下的指令:
1 | |
这里注释的部分是看是否添加了index.ndx文件来指定分组信息,如果有的话则需要将index.ndx也分配给各个文件夹。
不得不说gromacs的任务提交也算是个麻烦的事儿,根据计算核心的调整,计算内容的不同,计算脚本也是有很大的改变,以后有机会写一个自动化的脚本出来,至少也要让参数的改变变得更简单。
文件分配之后就可以开始运行计算了,依次切换到目录下先生成tpr文件,然后提交任务,代码如下:
1 | |
可以看到后面在加了热浴之后我用的mdrun的后续指令有所改变,这个是参考的这篇帖子:
GROMACS教程:GROMACS模拟空间非均相体系(板块结构)的并行性能:区域分解与PME节点设置|Jerkwin
这样设置的目录一来是为了节约计算资源,均衡负载,提高计算速度,二来是防止报错,因为此前我计算的时候出现过错误,就是因为计算核心的分配问题导致的,所以这里就继续加上了这个设置,不过这个数字的分配是根据核心的数量来变动的,对于帖子中的32核,那么计算运行的指令为:
1 | |
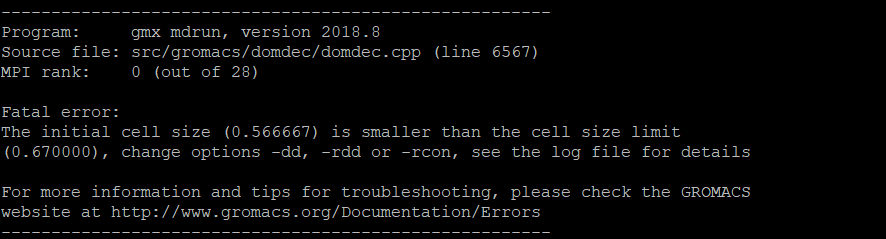
而我这里仅28核,一开始我是减少了npme分配的核心数量,将其但是这样分配会导致成品模拟的时候报错,以下是我开始用的指令:
1 | |
以下是报错提醒:
1 | |
随后我尝试着删去后面的区域分解的核心配置,用如下的指令进行计算,但是这个在一开始就提醒会导致巨大的性能损失:
1 | |
这个在计算的时候会出现如下的提醒:
1 | |
这样计算1ns的时候,效率如下:
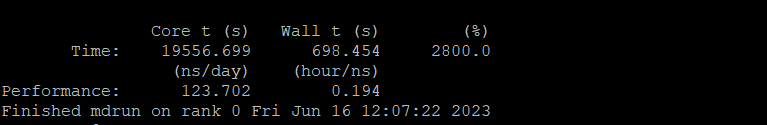
而后我尝试重新调整-dd 后面的区域分解计算核心数量,调整为如下的指令:
1 | |
这样计算的效率能够达到最好,这样计算上面相同的模型跑1ns的结果如下:
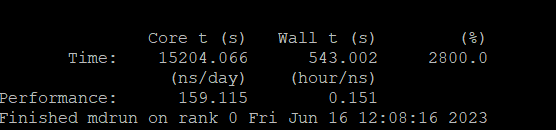
可以看到效率有一个显著的提升。
此外如果需要添加index.ndx,那么在每一个gmx grompp的后面都添加一个-n index.ndx即可,如下所以:
1 | |
最后放一下汇总的文件,我将其命名为gromacs.sh:
1 | |
这样的脚本写好之后就可以用sbatch指令提交了:
1 | |
这样就可以开始计算了,随后就是等计算完成再进行数据分析了,后续我看再写一篇关于这方面数据处理的博客吧。
总结
整个过程下来也算是一波三折,主要是换到了一个新的服务器上进行这个计算,导致了很多意想不到的问题,不过最后还是任务跑起来了,如果有报错的话可以多看看我的其它博客,我在计算过程中遇到的报错一般都会记录下来,而我这里全过程,如果有些步骤你无法理解,那么就照猫画虎的做吧,因为可能有些莫名奇妙的地方,就是为了规避某一个折磨了我好久的报错。