Pymatgen的学习以及应用
Pymatgen对于材料计算来说是一个非常有用的库,我前段时间基于这个库写了一个有关层状氧化物生成POSCAR的脚本,具体来说是使用了里面对Ewald能量的求解来帮我初步筛选一些能量,不过这个库的确是可以非常便捷的帮我编辑结构。
由于对这个库的使用一部分上涉及到了我课题的核心部分,所以代码肯定是不能全部放出来了,但是我会放出部分代码来作为教学,记录一下,此外也会涉及一些python语言的教程,一切的内容都是为了让初学者能够看懂我的代码。第一次写这种代码类型的教程,我也不是科班出身,很多内容都是我自己的理解,如有错误,还望海涵。
Python基础
我这里写的代码基础都是用的定义函数这一级别,还没到用到Class来定义对象,这样一定程度上会导致一些代码的冗余,但是好处就是我不用去回忆对象的一些定义了。
配置环境
环境的配置说的高大上,其实就是需要安装一下我们需要用到的库罢了,作为一个菜鸟,我还是习惯一开始就用的pip指令来安装python库,但是网上很多人其实不推荐这个方法,这个也好理解,因为pip安装的话,我的理解就是它是全局的安装的,也就是安装了这个库之后你在任意的目录下都可以直接引用这个库,这个听上去还不错,但是在实际应用的过程中这个就可能会导致一些莫名其妙的错误,比如有时候你写的code里面根本就没有使用这个库,但是它却跑出来打乱了原本认为的代码功能,所以Anaconda以及轻量级的Conda功能就是为了避免这样的环境紊乱导致的代码运行错误,它们会为你当前所需要运行调试的代码单独创建一个环境,这个环境里面只会有你所需要的库,不需要的库并不会被添加到环境中,这个会避免许多不必要的bug。
但是我用到现在位置,使用pip安装并没有遇到什么因为环境问题导致的bug,其实我觉得对于我们这种不是专业码农的人来说,pip应该也是够了,虽然传言它会导致这样那样的问题,但是实际上作为一个非计算机专业的人,我们可能很长时间都不会用到太多的库,比如我现在,为了完成这个课题,总共也就用了两个需要安装的库,一个是numpy,另外一个就是pymatgen,基本不用担心环境的问题,而pip无语伦比的优点就是,只要你有python,那么pip也就能用了,不需要再头大的去研究一下Anaconda或者Conda怎么安装。
啰嗦了好多,接下来说受pip安装pymatgen,这里需要看一下你的电脑上是不是有两个版本的python,比如我的服务器上就是既有python2.7,也有python3.5,所以如果我直接输入下面的代码:
那么这个时候调用的是python2的pip,安装貌似会出错,这种情况下一般来说就是需要用pip3的指令来安装,类似于python3.5的运行需要敲python3才能调用python3.5一样:
当然,如果你的电脑上只有一个python3以上的版本,那么直接用pip指令安装即可,我的windows电脑上就是这样安装的。
一些基础函数
首先是import函数,如下:
1 2 3 import warningsimport numpy as npfrom pymatgen.core.structure import Structure
我们要使用pymatgen这个库,自然是需要引用这个库的,import的作用就是将一些库导入,以便后续的使用,当然,这个函数也可以用来导入一些你自己写好的py文件,不过我这里不涉及这些,就不详细说明了。如上面的演示,import的使用有多种方法,简单粗暴的理解就是,最后的字符就是当你要使用引用的库时应该写的代码,比如我们导入了warnings这个库,并没有用as来重命名这个库,那么我们要用warning的时候就可以直接warnings.catch_warnings()这样来使用,而用了as,那么就是重命名了,比如这里要用numpy库里面的函数的时候,就需要用np.array()这样的形式。对于最后的from … import …这样的使用,就是对于pymatgen这样的大型库,里面的内容非常多,功能非常全面,而我们如果直接将整个库导入,一来会导致代码部分非常冗余,二来会导致运行过程中内存占用过大,所以我们这里可以从中选定一些特定功能的函数来导入,比如这里就是导入了Pymatgen里面的Structure类,使用的时候直接用Structure.copy()这样的形式就可以便捷的调用。
接下来就是def函数了,定义一个函数:
这个好理解,就是定义一个函数,相当于把一些功能封装好,便于我们后续调用这些功能。
代码格式
python的代码其实都是有一个固定格式的,我记得我之前学相关课程的时候有讲过为啥要这么写,但是我忘了,hhh,不过写代码以来还是时常感叹这样的代码格式的确可以让你的代码看上去非常的整洁,而且debug起来也非常的方便,后续添加函数或者屏蔽函数之类的也非常方便,以下是对这种格式的简单描述:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 import func..def func1 ():pass def func2 ():pass def main ():if __name__ == '__main__' :
这里一开始我们就是导入代码需要的库,然后就可以开始将我们需要的功能进行封装,比如读取POSCAR文件并提取指定原子的坐标,在指定坐标的位置上插入特定的原子,求解新的原子的Ewald能量等,写好这些代码之后,我们就可以依次调用这些函数,将它们写在main()函数中,最后再运行main()函数就行了,这样写的好处就是,我们前面定义功能函数的时候是无需在意顺序的,而且当我们需要停止使用某一个功能函数的时候,只需要再main()函数中将调用这个函数的代码行注释掉即可,如果需要添加什么函数也可以直接再main()函数里面添加即可。至于为啥要if __name__ == ‘__main__’:,我也不知道,搜过也没咋看懂。
代码的目标以及实现思路
目标
这里为了方便讲解以及练手,我就根据我自己做的课题,从中抽取部分内容来作为目标吧,如下:
♾️读取层状氧化物的POSCAR文件,在碱金属层中随机插入一定数量的原子,再求解它的Ewald能量,最后输出插入之后的POSCAR文件。
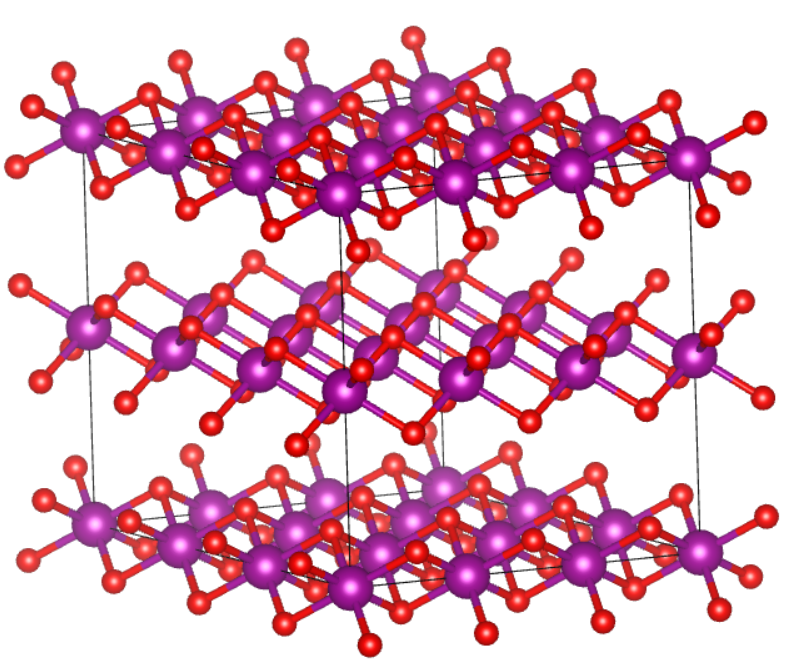
对于钠离子电池正极材料中的层状氧化物来说,过渡金属层一般由Mn,Ni这些元素构成,当然现在也有很多人开始用其它金属掺杂替换,我们这里也就不多做考虑。所以这里就是选择一各NiMn层状氧化物的基底材料,然后碱金属层添加Na原子,最后再计算它的Ewald能量并输出POSCAR,这个步骤我们可以得到一个粗糙模型的Ewald能量,这个理论上来说可以提前辅助我们判断结构的合理性。
实现思路
不难看出,整体的的工作可以分为四个功能模块,首先是读取文件,然后是插入原子,求解Ewald能量,最后是输出文件。也就是说我们需要定义四个函数,然后分别实现上面的功能。不过在Pymatgen中,对文件的读取还是很简单,这部分不需要单独写一个函数,而有关插入原子这一部分,比较重要的一点就是插入原子的位点怎么确定,为了进一步练手吧,我们就可以提取部分Mn原子的坐标,然后以他们的xy坐标,再设定一下z坐标来作为插入原子的点位坐标。
代码的实现
模型
这里关于模型的获取很简单,就是找一个常规的Mn基层状氧化物,然后将层间的Na离子删去,再保存为POSCAR文件即可,以下是我用的一个材料模型:
POSCAR的内容如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 test.xsd
代码的基本框架
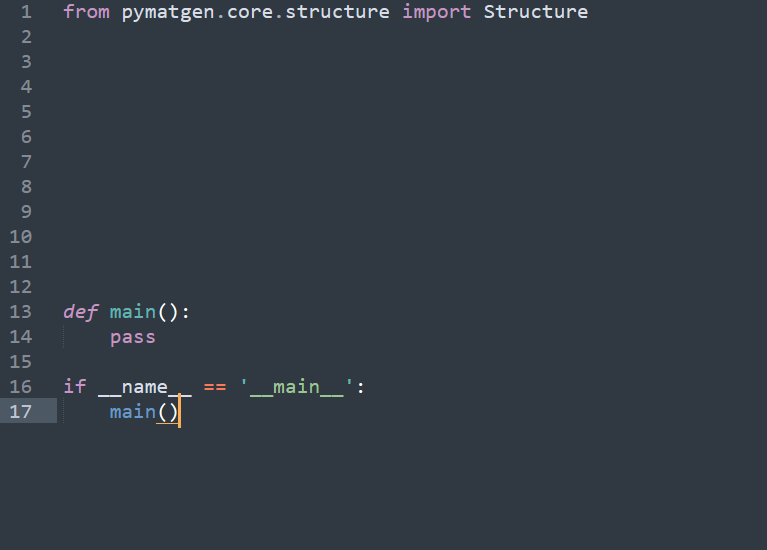
这里先搭建一个代码的基本框架,如下图:
说是框架其实就是一个壳子罢了,开始写代码之前一般都是这样先写着,我这里的
1 from pymatgen.core.structure import Structure
是因为Structure这个库后面肯定会用到,然后空白的区域就是我们需要自己写的函数去填充了,然后再在main函数中使用我们写好的函数。
读取模型文件并获得坐标
先上我这一步思路下写好的代码吧:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 from pymatgen.core.structure import Structurefrom pymatgen.io.vasp import Poscardef get_site (structure, element ):for site in structure if site.specie.symbol == element]for site in mn_sites]2 ] for site in mn_sites]1 + i if i <= 0.25 else i for i in mn_z_coords]for i in mn_z_coords if i > 0.75 ]for i in mn_z_coords if i <= 0.75 ]for i, z_coord in enumerate (mn_z_coords) if z_coord > 0.75 ]for i, z_coord in enumerate (mn_z_coords) if z_coord <= 0.75 ]sum (mn_layer1) / len (mn_layer1)sum (mn_layer2) / len (mn_layer2)2 1 ) / 2 print (Na_layer1_z)print (Na_layer2_z)for add_site in mn_layer1_site:0 ], add_site[1 ], Na_layer2_z]for add_site in mn_layer2_site:0 ], add_site[1 ], Na_layer1_z]print (len (site_coord))return site_coorddef main ():"POSCAR" "Mn" )if __name__ == '__main__' :
接下来一步一步的再解释一下代码吧,这里我们新导入了pymatgen的Poscar类,这是vasp类下的一个分支,可以帮助我们读取POSCAR文件,所以我们在调用我们新写的get_site()函数之前,先有一个读取文件并转化为structure对象的一个过程,这个过程没有写进函数内部的原因是我们后续很多操作都需要这个structure对象,为了节约资源,避免程序冗余,所以我就将它写到了main()函数中,然后将structure对象来传递给其它函数。
随后是函数内部,其实获取Mn原子的坐标在最开始的三行代码就实现了,后面的都是为了确定我们可以直接使用的坐标列表,毕竟我们不能直接将Na插入到Mn原子的位置上,而是与Mn原子对应的层间,在常规的层状氧化物中,有一部分Na离子在层间的位置就是与Mn原子对应的,也就是你从c轴方向看xy平面,Na离子与Mn原子之间的位置是重合的,它们空间位置的差异来源于z轴坐标,所以我们这个层间的位置x和y轴坐标就可以直接参考Mn原子的x和y轴坐标,但是z轴坐标就需要重新计算得到。
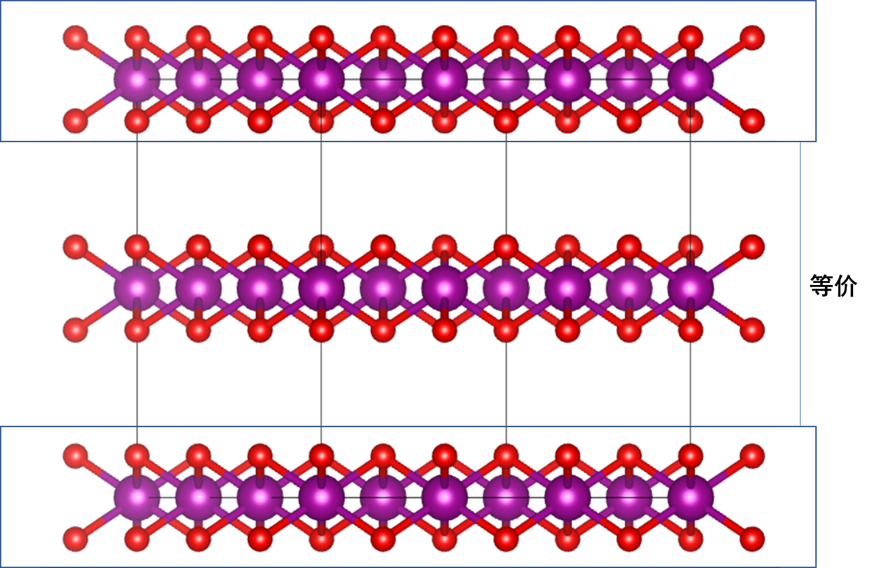
这里回头来再看一下层状氧化物的模型,虽然模型上有三层,但是实际上是只有两层的Mn原子的,三层是周期性结构显示的原因:
而观察POSCAR的内容也不难发现,一部分的z轴数据在0.5附近,而另外一部分则在1.0附近,但是没有大于1的数值,反而有一些负数,这些负数其实就是周期性结构中大于1的Mn原子。所以为了方便操作我们先对这个进行了归一化处理,也就是对它们+1,对于周期性结构的分数坐标来说,这个坐标信息是等价的,然后对这两层的原子进行了分层处理,将它们分管到不同的列表中。
然后我们也就可以根据这两层的数据来计算层间Na离子的z轴坐标。计算得到合理的z轴坐标之后,就是将计算得到了两层z坐标之后,就可以将Mn原子的xy轴坐标与计算得到的z轴坐标拼接起来,凑成一个新的坐标并保存到一个列表中,这里我为了确保两层的坐标数量一致且没有坐标重复的,所以根据两层Mn来分配了z轴坐标,比如对于layer1的Mn原子的xy坐标,与它们拼接的就是计算得到的layer2_z的数据。
最后我在里面提供了三个调试的函数,就是使用print来输出数据验证我们的代码是否按照我们的想法运行了,如果说代码没有错的话,前两个print打印出来的数据应该是两个小于1的数字,而最后一个print打印的数字则应该与Mn原子的数量对应:
随机采样坐标并插入Na原子并生成结构
接下来就是随机采样坐标了,我们这里设定嵌入3个Na原子吧,然后生成10个结构,这里代码就只放一个函数的了,不然就太长了,如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 import randomdef gen_structures (site_coord, structure, stuctreu_num, ele_num ):for i in range (stuctreu_num):3 )for random_sites in site_combine:for coord in random_sites:0 , "Na" , coord)print (len (new_structures))return new_structuresdef main ():"POSCAR" "Mn" )10 , 3 )
经过上一步获得了坐标信息,将其作为参数输入,然后还有10个结构数量和3个Na原子数量作为参数传入函数中,这里由于是随机采样,所以一开始还需要导入一下random这个库,我这里通过random.sample()指令来随机采样,然后采样了十次,不过我这里可能会存在一些采样结果是一致的,这里就没有做重复筛选的功能了。
然后就是根据采样的结果,遍历之后获得坐标信息,然后在编号为0的位置插入Na原子,再将插入之后的新结构保存到列表中返回。
这里的调试如果运行成功的话,返回的是我们最终生成的结构数量,我们设定的是10个,那么这里也应该打印10。
计算Ewald能量并输出结果
这一步就是按部就班的根据得到的结构然后计算Ewal能量了,难点就是理解Ewald能量的计算方法以及pymatgen中的应用,理论知识就不多赘述,直接上应用吧:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 from pymatgen.analysis.ewald import EwaldSummationdef get_ewald_energy (new_structures ):"Na" : 1 , "O" : -2 , "Mn" : 4 }0 for structure in new_structures:1 "poscar" , filename=str (index_n) + "_POSCAR" )sorted (file_energy_list, key=lambda x: x[1 ])for i in sorted_energy_list:print ("结构ID:" + str (i[0 ]) + " 的EWald能量为" + str (i[1 ]))def main ():"POSCAR" "Mn" )10 , 3 )
要求解Ewald能量就需要用到pymatgen中的EwaldSummation类,所以需要导入这个类,然后将之前得到的new_structures列表传递给函数,函数内遍历这个列表,获取每个结构,给每个结构添加价态,然后就是求解Ewald能量,求解之后按照编号+能量的方法来组成一个新的列表并将每个结构输出为POSCAR文件,最后还会打印每个结构的Ewald能量。
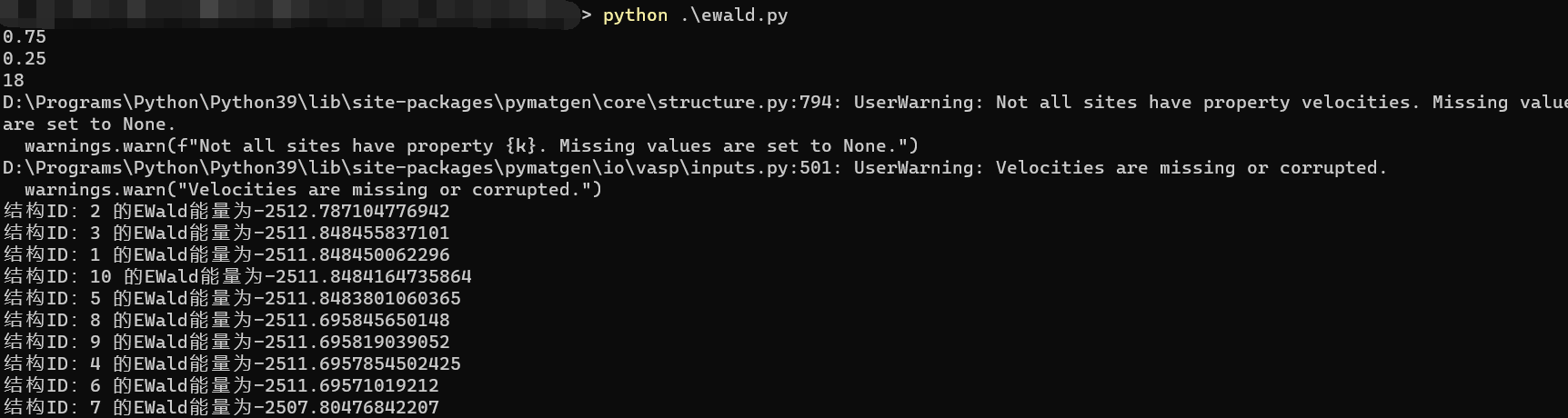
到了这一步,运行之后的结果如下:
中间有一些warning,这是无害的,如果看不惯可以考虑导入warning库来忽略这些警报,同时这个脚本所在的目录下也会多出来10个POSCAR格式的文件,并且按照1-10的编号命名好了的,可以自行查看。
总结
代码总览
最终写好的代码如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 from pymatgen.io.vasp import Poscarimport randomfrom pymatgen.analysis.ewald import EwaldSummationdef get_site (structure, element ):for site in structure if site.specie.symbol == "Mn" ]for site in mn_sites]2 ] for site in mn_sites]1 + i if i <= 0.25 else i for i in mn_z_coords]for i in mn_z_coords if i > 0.75 ]for i in mn_z_coords if i <= 0.75 ]for i, z_coord in enumerate (mn_z_coords) if z_coord > 0.75 ]for i, z_coord in enumerate (mn_z_coords) if z_coord <= 0.75 ]sum (mn_layer1) / len (mn_layer1)sum (mn_layer2) / len (mn_layer2)2 1 ) / 2 print (Na_layer1_z)print (Na_layer2_z)for add_site in mn_layer1_site:0 ], add_site[1 ], Na_layer2_z]for add_site in mn_layer2_site:0 ], add_site[1 ], Na_layer1_z]print (len (site_coord))return site_coorddef gen_structures (site_coord, structure, stuctreu_num, ele_num ):for i in range (stuctreu_num):3 )for random_sites in site_combine:for coord in random_sites:0 , "Na" , coord)return new_structuresdef get_ewald_energy (new_structures ):"Na" : 1 , "O" : -2 , "Mn" : 4 }0 for structure in new_structures:1 "poscar" , filename=str (index_n) + "_POSCAR" )sorted (file_energy_list, key=lambda x: x[1 ])for i in sorted_energy_list:print ("结构ID:" + str (i[0 ]) + " 的EWald能量为" + str (i[1 ]))def main ():"POSCAR" "Mn" )10 , 3 )if __name__ == '__main__' :