计算溶液的配位环境
继之前得到乙腈分子的拓扑文件之后,进一步的我就要开始模拟计算溶剂的配位环境了,这里就把教程继续写下来吧,越写教程肯定是会的越多的,这篇博客的目的主要是为了记录下这次计算的全过程,所以我是从开始写文件的同时也开始写这篇博客,力保整篇博客能够完全囊括从初期准备到计算数据后处理全过程的内容,同时有什么报错也会写进来并把解决办法一并附上。
top、itp文件的编写
之前在学习的时候就讲过,我们在写top文件和itp文件的时候就要有意识的将它们分门别类的写好,现在我要模拟的体系是1 M Zn(OTf)2 溶在乙腈中,含有8%的水,所以我们需要Zn、OTf、AN、Water的itp文件,然后就是一个nonbonded.itp文件和一个topol.top文件。
前面这四个itp文件根据我之前的学习内容,其实都已经有了,这里我就都贴出来:
Zn.itp,这个我用的是Amber03力场里面的参数:
1 2 3 4 5 6 7 [ moleculetype ]
CF3SO3.itp,这个是我根据参考文献写的,写的相当冗余:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 [ moleculetype ]
AN.itp,这个就是根据前面的博客得到的:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 [ moleculetype ]
water.itp,这个是是我在网上找的OPC水模型:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 [ moleculetype ]
为了便于后面编写nonbonded.itp文件不重复,所以我都是将[ atomtypes ]去掉了的。
接下来就是写nonbonded.itp了,这个就是将之前这些各个itp文件的[ atomtypes ]整合到一起就行了,我这个体系的nonbonded.itp如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 [ defaults ]
这里突然想到一个网站,之前我在参考文献写CF3SO3的itp文件的时候,文献中给出的ε单位是K,而gromacs的单位是KJ/mol,然后我最开始没注意这个单位换算,算出来的结果就不准确,然后我是自己去查各种参数,最后自己在Excel里面算出来的单位转化之后的值,相当的麻烦,后面我反而是在用Lammps的时候,为了找势函数而找到了一个能量转化的网址,非常好用:
Energy Converter (colby.edu)
可以直接用这个网站来进行单位转化。
最后就是topol.top文件了,这个的编写先需要知道我们在conf.gro中的分子数量以及顺序,但是我这里就先大致算了一下原子数量,然后取整数,先把topol.top文件写好之后再按照top文件里面的数量和顺序生成盒子。以下是我用excel大概算了一下4.5nm的正方体盒子所需的各个分子数量:
比较乱,大概看一下就行,需要1100个乙腈分子,60个Zn(OTf)2以及200个水分子,这里8%我用的是质量分数并且没有计算盐的质量。
接下来就是按照这个数量去编写top文件了,以下是我写的topol.top文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 #include "nonbonded.itp"
以上就是所有的相关的拓扑文件了,写好之后可以进行下一步了。
盒子的生成,产生conf.gro文件
这里大部分模型我们之前都有的,CF3SO3在之前的学习中我是通过Gaussian优化之后的,Zn离子更是简单,直接画一个原子即可,水模型则是在Gromacs的安装目录下找一个四点水模型即可。乙腈分子的模型我们也是通过Gaussian优化之后可以获得。这样目录下应该有这四个pdb文件:
接下来将这四个文件上传到服务器中,用packmol来生成盒子的pdb文件,packmol的安装这里就不多做赘述,可以自己搜一下,很简单的一个小软件,但是功能非常强大。用packmol需要写一个.inp文件作为参数文件,即告诉这个软件你要建一个多大的盒子,放多少个分子,放哪些分子,以下是我写的mix.inp文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 tolerance 2.0
这个文件的具体参数可以看packmol的操作手册,还是很简单的,我的这个mix.inp就是按照之前写topol.top文件里面的分子数量以及顺序写的,然后盒子大小就是4.5nm,然后会将生成好的盒子保存为box.pdb。然后在命令行里面输入:
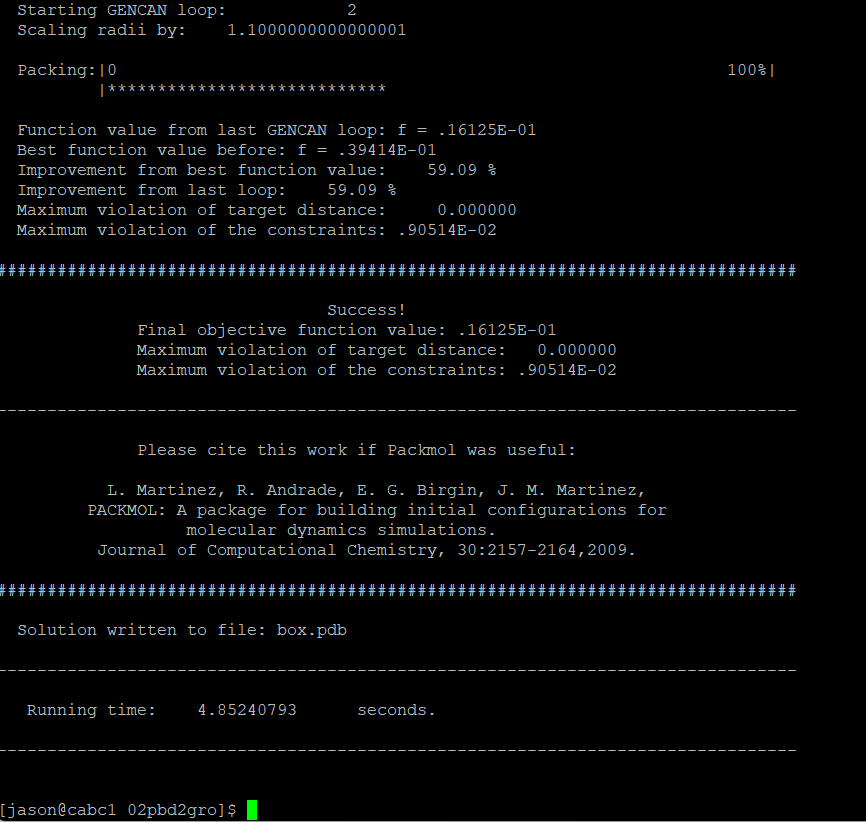
以下是运行完成后的截图:
生成之后就可以进一步用gmx的指令将其转化为gro格式的文件,在命令行输入:
1 gmx editconf -f box.pdb -o conf.gro
这里运行成功后不同版本呈现的页面会不一致,但是gromacs运行成功后末尾一般都是被一句名言,如图:
这样目录下就会有生成的conf.gro文件了。
tpr文件的生成并提交计算任务
有了top文件和gro文件我们就可以开始生成tpr文件了,这也是计算前的最后一步了,在这之前我们需要先编写mdp文件,也就是参数文件,告诉Gromacs你怎么计算,我按照之前的学习内容,这里也采用5步计算方法,就是2步能量最小化,2步预平衡加上最后的成品模拟。以下是每一步的mdp文件,具体的参数解释这里就不赘述了,有不懂的可以参考Gromacs的手册。
min.mdp:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 define = -DFLEXIBLE
min2.mdp:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 integrator = steep
eql.mdp:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 integrator = md yes
eql2.mdp
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 integrator = md yes
prd.mdp:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 integrator = md yes
这里我先以min.mdp文件为例来生成tpr文件,确定top文件跟gro文件没有什么问题了,再用一次性连续运行5次任务的脚本提交任务。
将top相关文件,gro文件以及min.mdp放在同一个目录下,在命令行中输入:
1 gmx grompp -f min -o min -pp min -po min
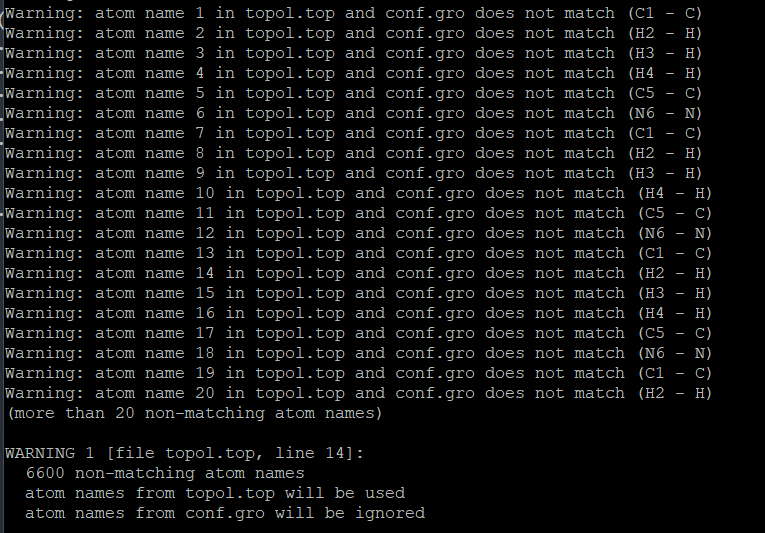
这个时候出现了Warning,如下图:
这个警告一般是top文件里面对原子的命名与gro文件中的名字不匹配导致的,修改起来也很简单,将对应itp文件里面的名字改成跟gro文件里面的一致就行,比如我这个很明显是乙腈的拓扑文件有问题,那么就将乙腈的AN.itp里面的[ atoms ]改一下,以下是修改之前的,也就是我前文中放的:
1 2 3 4 5 6 7 8 [ atoms ]type residue resname atom cgnr charge mass
修改之后的为:
1 2 3 4 5 6 7 8 [ atoms ]type residue resname atom cgnr charge mass
将atom那一列跟gro中的标记对上即可。
修改之后再执行之前的命令就没有啥问题了,成功生成了min.tpr文件:
那么接下来就可以正式开始run了,我贴一下我写的一个小脚本,它可以在一个节点上连续提交任务,就是能够在第一次能量最小化之后自动开始下一次能量最小化,再自动开始预平衡,知道最后成品模拟完成,这个其实在我之前的博客里面也有,但是这篇博客就是为了完整,所以也贴出来吧。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 #!/bin/bash source /soft/profile.d/gromacs514.shif [ ! -d "1min/" ];then mkdir 1minfi cp min.mdp conf.gro *.top *.itp ./1mincd 1mincd ..if [ ! -d "2min/" ];then mkdir 2minfi cp ./1min/min.top ./2min/cp ./1min/min.gro ./2min/cp min2.mdp ./2min/cd 2mincd ..if [ ! -d "3eql/" ];then mkdir 3eqlfi cp ./2min/min2.top ./3eql/cp ./2min/min2.gro ./3eql/cp eql.mdp ./3eql/cd 3eqlcd ..if [ ! -d "4eql/" ];then mkdir 4eqlfi cp ./3eql/eql.top ./4eql/cp ./3eql/eql.gro ./4eql/cp eql2.mdp ./4eql/cd 4eqlcd ..if [ ! -d "5prd/" ];then mkdir 5prdfi cp ./4eql/eql2.top ./5prd/cp ./4eql/eql2.gro ./5prd/cp prd.mdp ./5prd/cd 5prd
这个脚本就是把它当作任务提交的脚本就行了,比如我的服务器集群是用sbatch提交任务,那么我们就可以直接在工作目录下,将这个脚本保存为gromacs.sh ,然后将之前生成tpr所需的top、itp、gro以及那5个mdp文件复制到工作目录下,最后在命令行输入:
这样就可以提交任务自动开始计算了,这个提交任务之后会依次在工作目录下创建五个步骤的目录,方便我们后续自行查看计算进度并按照需求提取数据,运行之后的工作目录如下:
结果的提取—径向分布函数以及配位数
这次做这个MD模拟的主要目的还是为了溶液中溶质的配位环境,所以需要导出径向分布函数以及配位数。计算结束后目录下会有xtc格式的文件,我们就可以从中提取我们需要的数据,gmx提供了相当便捷的脚本来帮我们提取数据,在提取数据之前,需要新建一个索引文件,方便我们按照自己的需求来提取数据,先输入以下的指令:
1 gmx make_ndx -f prd.gro -o index.ndx
然后会进入到一个操作界面,如下图:
其实如果前面我们在写top文件和gro文件的时候能够注意一些细节的话,这里它自带的分组就应该够用了,但是很显然,我们这里有些分组还是不够用的。
我们现在是想要获取Zn离子周围的配位情况,所以以Zn离子肯定是需要单独分一个组的,这上面已经有了,然后是水分子中的OW,水分子中的HW,CFSO3中的O,F,乙腈中的N,H,尽量把能分出来的组都分出来吧,即依次输入以下指令:
1 2 3 4 5 6 7 a OW
最后的q就是为了保存index.ndx文件。
然后就可以输入以下的指令来生成径向分布函数文件以及配位数文件:
1 gmx rdf -f prd.xtc -n index.ndx -cn
这里后面的-cn就是代表输出径向分布函数文件的同时输出配位数文件,如果不需要配位数文件的话可以不加,想起来之前初学的时候我还自己写脚本来计算配位数,都是泪啊。
输入上面的指令也会进入到一个操作页面,需要我们选择参照物以及需要导出的分布信息,参照物这里就是选择Zn离子了,然后依次选择之前我们分类好的原子即可:
我这里就是先选择7,然后依次选13-18,最后Ctrl+D保存即可,这之后gmx会自动计算一会儿,理论上来说你模拟的时间越长这里花费的时间也就越多,不过一般也不会很长时间吧,计算之后目录下就会多出来两个文件了,用这两个数据文件画图即可。
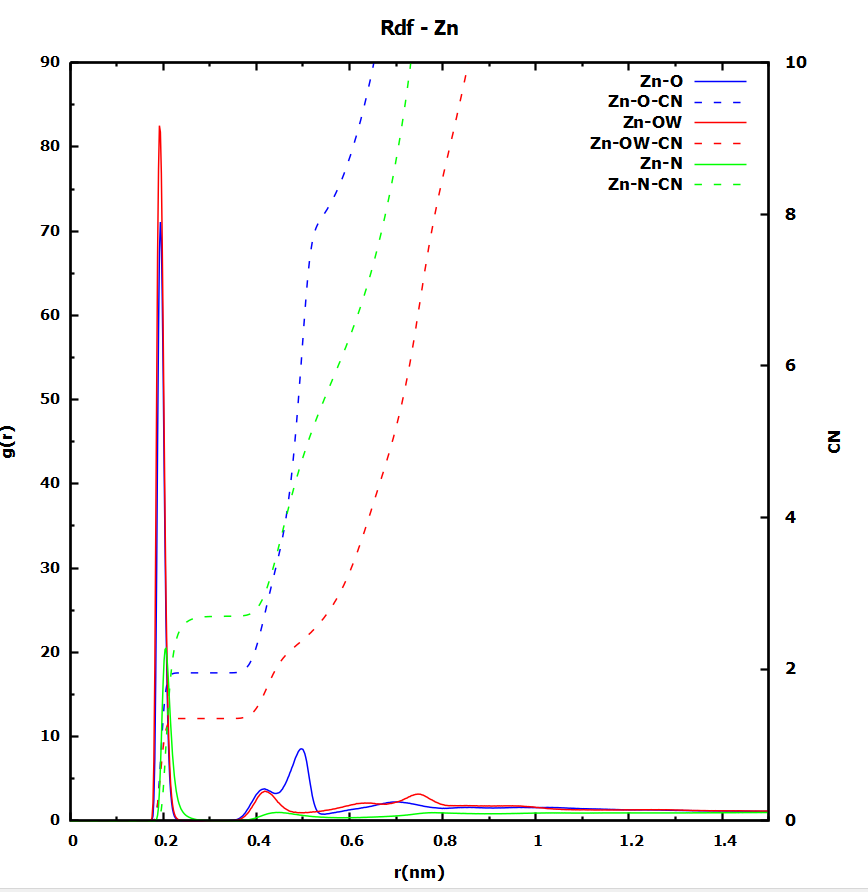
我使用win版的gnuplot画的图,画出来的图如下:
当然,这里也可以导出轨迹文件在VMD里面查看,在工作目录下输入:
1 2 gmx trjconv -f prd.xtc -s prd.tpr -pbc mol -o prd-mol.xtc
然后选择全部原子,输出一个prd-mol.xtc,这个文件就会比较大,接下来将prd.gro和prd-mol.xtc输出到自己的电脑上,打开VMD,用cd指令切换到这两个文件所在的目录,然后输入
即可以看到它的轨迹了,如下图:
以上就是全部的工作了。
总结
总的来说,这整个模拟下来涉及到的软件还是相当的多的,而且要各种倒腾文件,还是比较麻烦的,但是熟悉了之后还是没有很难以理解的步骤,大概就是建模-写拓扑文件-生成tpr-模拟-提取结果这样的一个步骤,比较麻烦的时每一个步骤基本都对应着一个新的软件环境,需要在不同的软件之间反复横跳,出错了往往就是重来。