乙腈分子力场参数的获取
最近计算一个体系要用到乙腈的模型,这里就想顺手在生成乙腈分子拓扑文件的时候记录一下,毕竟教程肯定不会嫌多的。
建模并优化—Gaussian
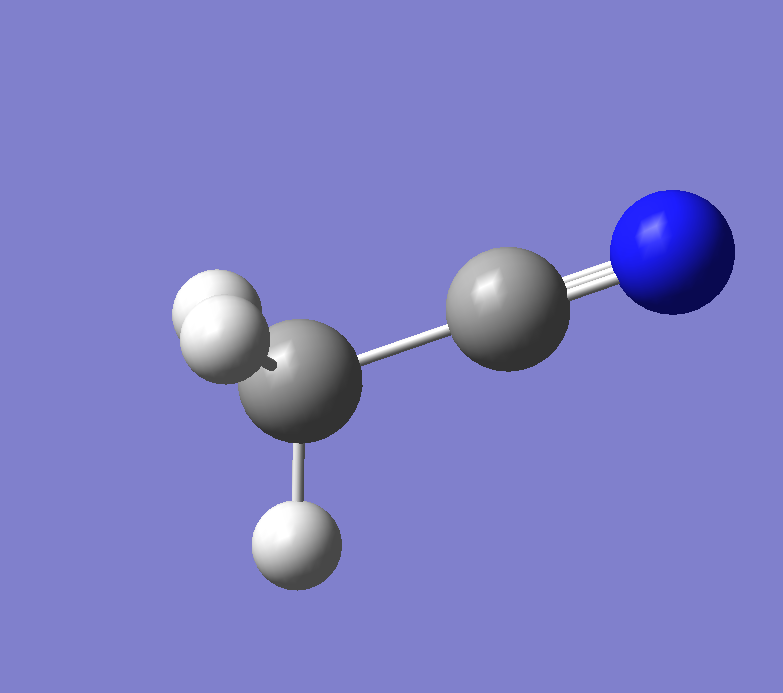
通过Gaussian先对建模的乙腈(AN)分子进行结构优化,并获取它的fchk文件,一下是建立的模型:
导出gjf格式的文件,然后修改一下gjf的文件内容,一定要记得加上freq关键词:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 %chk=./AN.chk
这个分子还是相对大一点,所以我是上传到服务器集群上计算的,优化完成之后用formchk转化一下格式,将chk格式的文件转化为fchk文件,在工作目录下输入:
注意,用formchk时需要注意你的环境变量里面是否添加了这个脚本,没有的话记得联系你们的管理员。
得到了fchk文件之后我们就可以开始下一步操作了。
Multiwfn生成电荷信息(RESP)
对于乙腈这样的分子结构来说,极性比较强,而且在我计算的体系中它是作为溶剂,所以获得每个原子的电荷再来编写它的拓扑文件是非常有必要的,Multiwfn提供了非常便捷的操作来生成分子的RESP电荷。
安装好Multiwfn之后,直接再命令行敲Multiwfn,然后输入./AN.fchk,将刚刚得到的fchk文件输入,再进行后续操作,一下是输入的内容:
1 2 3 4 5 6 Multiwfn
后续的操作在Gromacs的学习以及应用(五) - PhoenixJason 这篇博客中有解释,可以去看看。
这样我们会获得一个AN.chg文件,cat一下这个文件看一下:
1 2 3 4 5 6 7 [jason@cabc1 01Gaussian]$ cat AN.chg
这里面保存了AN每个原子的元素、坐标信息和所带电荷。
这样我们目录下就会有.fchk,.chg,.log文件,为了方便操作,接下来我们可以将这三个文件全部输出到自己的windows系统上进行下一步操作。
Sobtop生成带电荷信息的top、itp文件
拿到.log文件之后首先在GaussView中看一下计算的收敛情况,看有没有虚频之类的,一般这种小分子都不会出现什么大问题。然后就可以输出一份mol2文件作为Sobtop的输入文件。
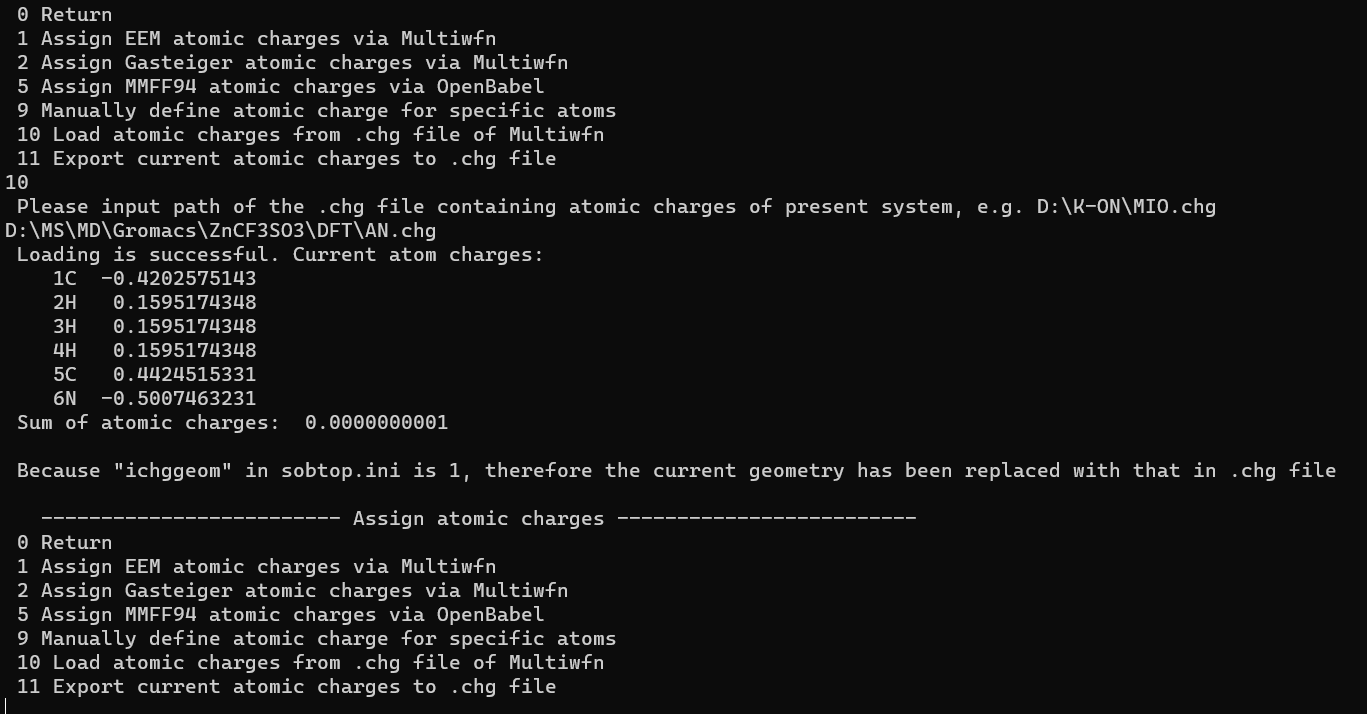
打开Win版的Sobtop,以刚刚输出的mol2文件作为结构文件输入(最简单的办法就是直接将那个文件拖到sobtop的命令行页面中),然后先选择7,为每个原子分配电荷,再选择10,用chg文件来分配,然后输入刚刚我们得到的chg文件的路径(也可以直接拖入),
成功分配之后如下图:
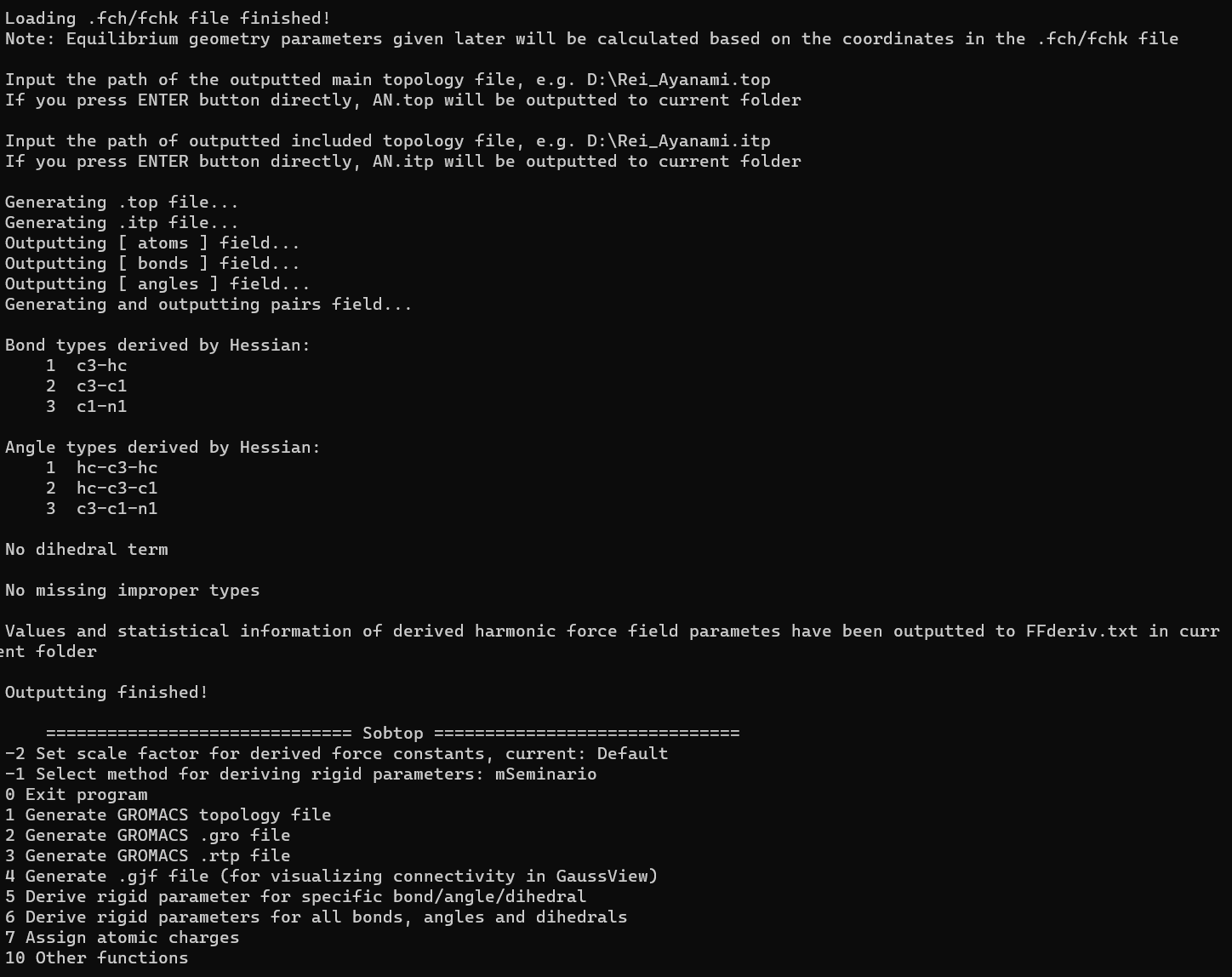
接下来就可以进行下一步,生成top文件了,再输入0,return到开始的界面,然后输入1生成top文件,这里我们用GAFF力场,输入3,所有原子都被指定了类型,那么就输入0,进入下一步,设定成键信息,因为我们有AN分子的fchk文件,所以这里我们可以直接选2,这是Sobtop的一种根据Gaussian计算结果获取成键参数的方法,然后拖入刚刚得到的fchk文件,最后就是连续两下Enter键,将得到的top文件和itp文件输出。
这里遇到一个蜜汁bug,之前还可以正常输出top和itp文件的,我这想截个图就会在输出文件的那一步直接关闭窗口,而且没有top文件的输出,很奇怪,试了好几遍都不行。
后面测试了一下,初步估计是sobtop目录下已经存在了AN.top和AN.itp文件导致的,而且不知道为啥,我尝试几遍之后fchk文件会变得不能用,原来300多kb的文件会变得只剩下1kb的大小,需要重新从服务器上下载,很奇怪,但是最后还是弄好了,一下是完成之后的截图:
这样我们即可以成功的获得AN分子的GAFF力场的拓扑文件了,接下来我把这俩文件的内容贴出来吧,需要的可以自取:
AN.top:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 ; Created by Sobtop (http://sobereva.com/soft/sobtop) Version 1.0(dev3.1) on 2023-04-18
AN.itp:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 ; Created by Sobtop (http://sobereva.com/soft/sobtop) Version 1.0(dev3.1) on 2023-04-18
总结
总的来收,整个力场文件的获取就是Gaussian —> Multiwfn —> Sobtop,后面两个软件Sobereva都有非常详细的教程,具体可以参考他的教程,这样应该是能够解决大部分小分子气体的力场参数的。