基于sobtop的Windows下获取小分子的top文件
基于sobtop的Windows下获取小分子的top文件
GSW 计算,获取chk,转化为fch文件
使用sobtop生成top文件
最近的一些工作涉及到类似于氮气,氢气这样的小气体分子的动力学模拟,所以需要得到这些小分子模型的力场参数,而最近课题组的服务器资源比较紧张,小分子优化也花费不了多少时间,所以我就想着直接用win版的高斯结合着sobtop来生成top文件了。
Gaussian优化小分子并获得chk文件
这里以为例进行演示,我们先建模,这个很容易,然后导出gjf格式的文件:
生成的gjf文件可以直接用sublime(或者记事本,任何能打开文本文档的都行)打开修改,这里我是因为使用了Linux版本的后遗症吧,习惯在gjf文件里面修改配置内容,其实win版的高斯可以有UI界面供修改计算的参数的。以下是我的gjf文件的内容,注意一定要打开freq计算,这个是为了后面生成top文件做准备:
1 | |
然后用Gaussian 16W打开(这是我的笔记本电脑上安装的版本),界面如下:
因为我们在gjf文件里面已经配置好了计算的参数,所以这里不需要修改什么内容,直接点右边的run即可,然后会跳转到保存out文件的选项,选择好目录保存即可。
这个计算很快就结束了,计算正常结束如图:
同时你设定的目录下应该会多出两个文件,分别为.out和.chk文件,那么就可以进行下一步了。
转化chk文件为fch文件
chk文件是一个二进制的文件,sobtop是没办法直接读取的,而通过Gaussian自带的formchk工具将chk文件转化为fch文件才能正常读取,这个操作我在之前的计算动力学直径的时候做过,不过那个是Linux版本的,这里win版的一个是转化生成的文件格式不一样,二一个是转化的方法也不一样。
首先我们需要找到Gaussian的安装目录,方法很简单,根据快捷方式,鼠标右键,打开目标所在目录即可,我的因为它是被我固定到了开始菜单栏,所以这个跳转要进行两次。
进到安装目录下,会有很多exe文件,找到formchk.exe文件
然后可以直接复制一份这个exe文件出来,这个formchk.exe应该是不存在路径依赖的一个小工具,你把它移到任意目录下都可以运行的,可以直接复制它到你保存chk文件的目录下,然后双击。
这里会弹出一个命令行页面,上面写着Checkpoint file?:
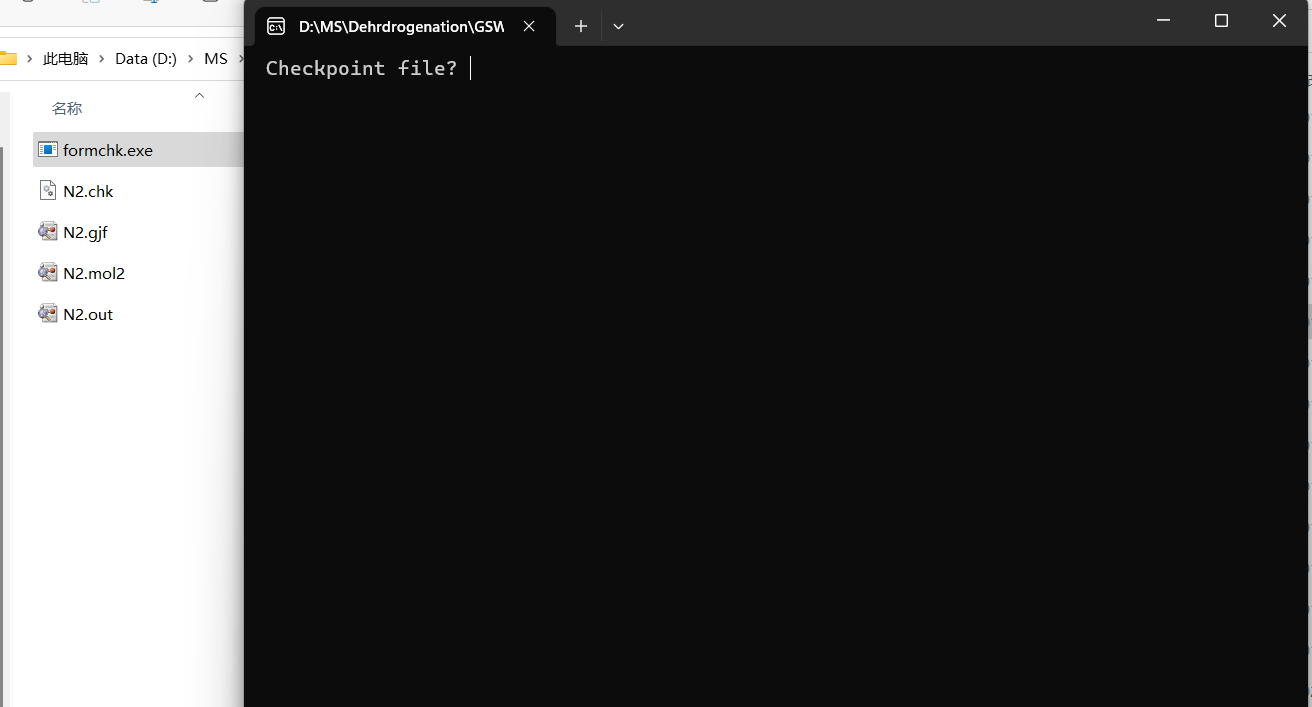
这里直接在后面输入当前目录下的chk文件的名字即可,比如我这里就直接输入N2.chk,然后回车键,然后目录下就会多出来一个N2.fch文件,这样就代表转化成功了,即可以开始下一步了。
此外还有就是mol2文件的问题,这里既然结构优化完成了,那么我们还需要用Gauss View读取一下out文件或者chk文件,查看一下优化有没有问题,看看有没有虚频,然后导出一个优化之后的mol2文件,用更准确的小分子结构文件进行后续的操作。
Sobtop生成top文件
接下来就是sobtop生成top文件了,找到sobtop所在的目录,双击sobtop.exe文件,然后在N2.mol2的目录下鼠标右键该文件,复制它的文件路径,粘贴到sobtop.exe运行后弹出的命令行页面中,输入N2的结构信息。
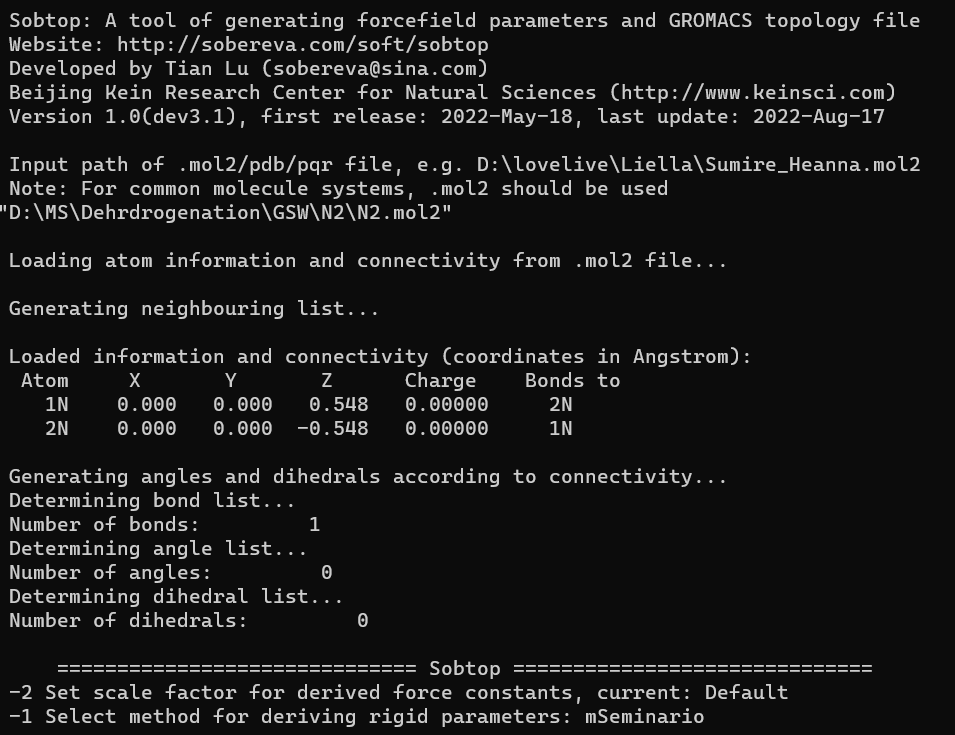
然后就可以根据它的提醒进行操作了,先选择1,生成Gromacs的top文件,然后选择2,根据GAFF力场为为所有原子分配类型,再然后选2,所有键都根据 mSeminario method来定义,这之后就会让你输出fch文件的路径,直接复制chk文件的路径过来即可,注意这里如果文件路径有双引号是不可以的,所以如果粘贴过来的文件路径有双引号,记得先去掉:
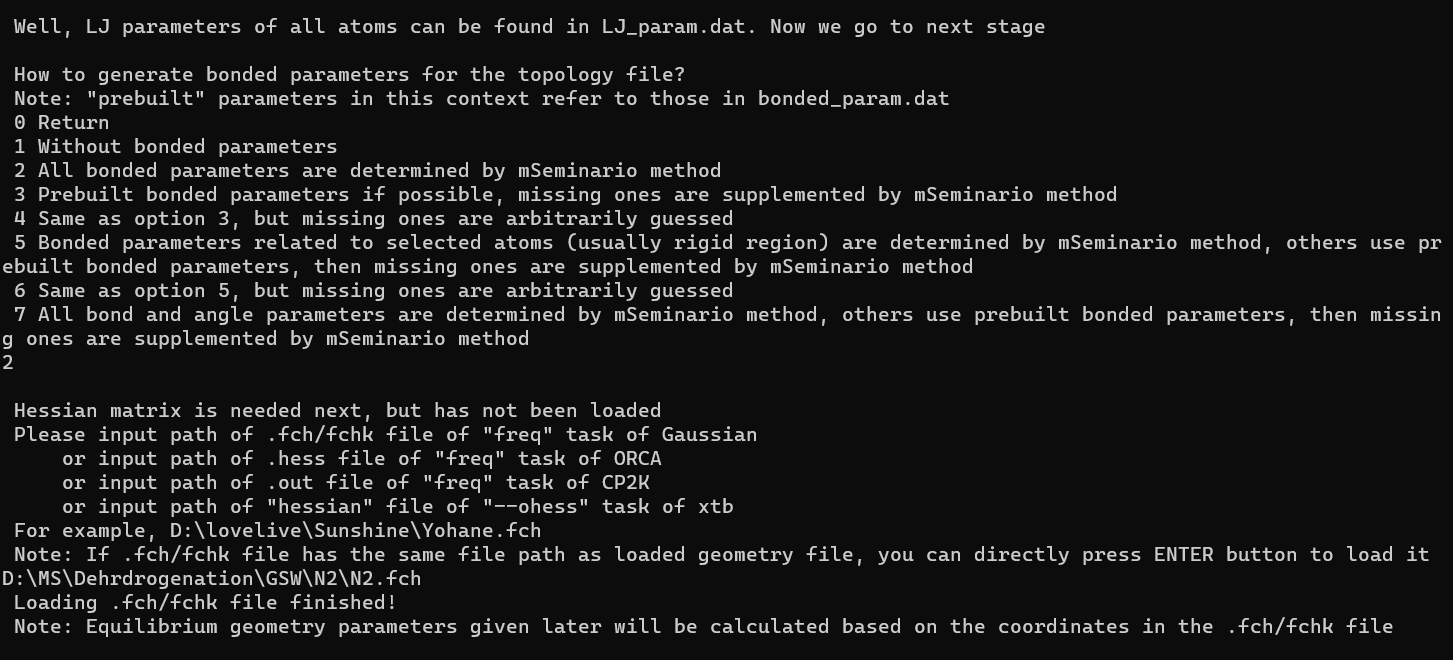
出现下面的那个loading …字符即代表这个加载成功了,然后就是top文件的输入,会生成两个文件,一个是top文件,还有一个itp文件,都需要你指定输出的路径,目前不清楚是这个软件的bug还是我这目录有啥问题,我尝试指定路径输出都不行,会直接结束程序,只有啥都不输入,直接回车用sobtop的目录输出,然后即可以在sobtop的目录下找到输出的文件。小有瑕疵,但是问题不大,以下是我生成的两个文件的内容:
N2.top
1 | |
N2.itp
1 | |
总结
小分子的top文件生成用sobtop还是相当方便的,在windows上操作也很方便,没有什么特别麻烦的地方,而且这样可以减少一步倒腾gjf文件到服务器上,在从服务器上倒腾fchk文件的过程,直接在window的工作环境下,后续对多个小分子的itp文件进行重写也很方便,感觉可以替代以前在Linux上生成top文件的操作了。