使用Gaussian来搜索过渡态
使用Gaussian来搜索过渡态
经过前端时间的爆肝工作后,现在终于可以挣得一些喘息的时间来完成自己的课题了,不过准备着手开始干了才发现我自己对于计算过渡态真的是一无所知,所以这里我打算还是继续学习一下使用Gaussian来计算搜索过渡态,还是通过复现文献的手段来实现吧。
参考文献是这一篇文章:
这是一篇纯计算的文章,感觉上对电解液添加剂的研发还是有一定的启发作用的,这里面对对过渡态的计算方法还是很适合电解液的开发的,毕竟用我老师的话来说,电池电解液体系是一个非常Dirty的体系,里面有太多的影响因素了,咱们对产物的推导往往很难凭借自己的化学知识推导出来,通过这种未知产物的计算来获取副反应产物是一个比较不错的方法。
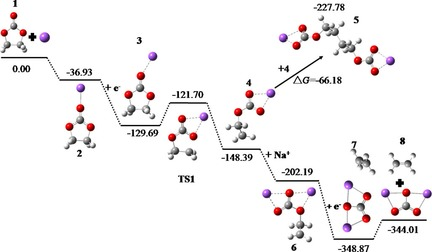
以上就是该文献对FEC在电解液中发生反应的过渡态计算,这里也把计算方法贴出来:

为了让自己能够真正学会使用Gaussian来计算过渡态,我这里就以上图对EC的过渡态计算为例进行复现,用自己的复现结果与文献对比,练手并思考着将其用在自己的课题上。
初期的结构优化
在过渡态开始计算之前肯定是不能直接用自己建立的模型进行计算的,还是要进行初步的结构优化,再进行下一步的过渡态计算。
不带电模型的计算
先建立一个EC的分子模型进行结构优化,再将优化结果导出,看是否有虚频,若没有虚频则再添加一个Na离子到模型中继续进行优化,这里的优化步骤就不多赘述,我结构优化采用的关键词如下:
1 | |
报错
其实在计算开始出现过报错:Error termination request processed by link 9999.对此我也查了一下原因,感觉造成这个报错的原因还是挺多的,首先就是需要检查自旋多重度是否正确,因为对于一些初学者来说这个还是会被忘记的,然后是检查结构是否合理,我这个报错最后就是重新调整了初始结构才没有继续报错(注意多使用GaussView自带的clean按钮)此外也可以试着在opt里面添加关键词cartesian,关于这个问题的处理可以参考Sobereva的帖子:
量子化学计算中帮助几何优化收敛的常用方法 - 思想家公社的门口:量子化学·分子模拟·二次元 (sobereva.com)
此外还遇到了这个报错:EpsInf not defined for this solvent.这里参考这篇帖子进行解决:
用eps加溶剂出错 - 量子化学 (Quantum Chemistry) - 计算化学公社 (keinsci.com)
简单来说就是再eps前面在加一行epsinf,由于我们这种计算并不会涉及到这个值,所以随便设定一个值就可以了。
中途经历了各种报错再尝试之后终于能够正常的收敛了,收敛速度还是挺快的。
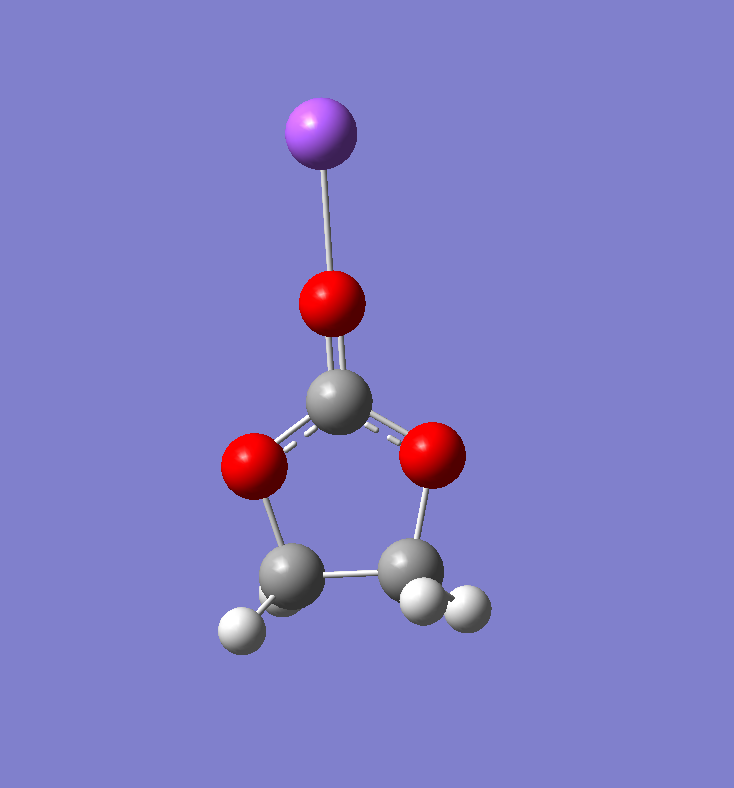
优化结果
结构优化后的结果如下:
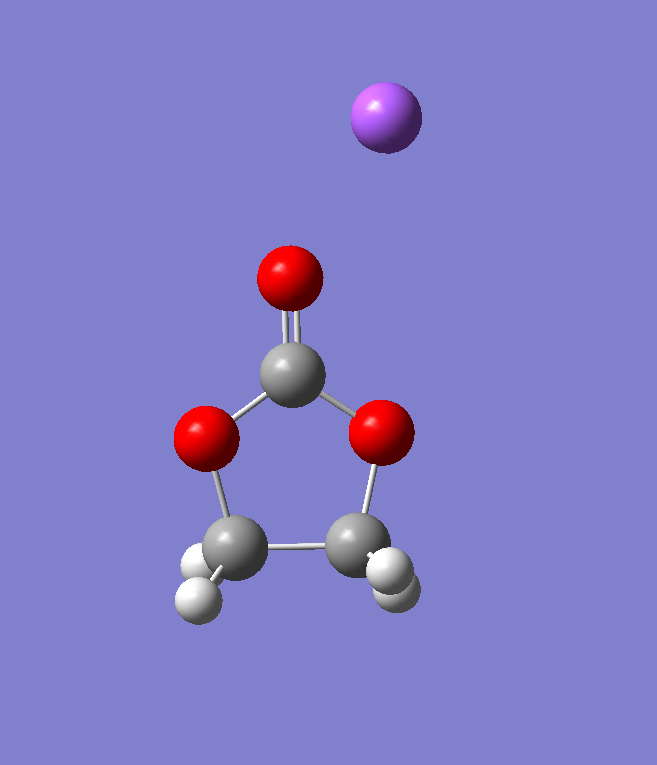
带一个电荷的模型的计算
上一步计算得到的结构跟文献中的结构还是挺像的,现在要进一步计算,先对它增加一个电荷,再次进行结构优化,这个加电荷的操作就比较简单了,直接修改一些gjf文件就行,注意这个时候自旋多重度也需要改成2(电子数量发生了变化),然后继续结构优化,优化出来的结果就能看到Na离子向一侧的氧原子有略微的偏移,这个与文献中的结果也能对应上。
过渡态计算
接下来就是进一步进行过渡态相关的计算了,我也是看了一些教程,Gaussian的过渡态计算其实也是有比较多的方法,与我之前了解的,给出产物与反应物,插值寻找过渡态的方法不同,用Gaussian计算最常用的还是提供一个初猜,再根据初猜的受力情况进行计算,再来寻找过渡态。这个方法就相较于前面我所想的想法优点在于可以节省不少计算量,也可以极大的减少工作量,不过缺点就是要自己对体系比较了解,对基本的化学知识需要有较深的理解。
获得初猜结构
不难看出,按照这种提供初猜的方法来找过渡态,首要的就是获得初猜,这里Gaussian也提供了一些方法来方便我们找到合适的初猜。在Gaussian中opt关键词modredundant可以让你固定一些键(或者角以及二面角)来优化其它的部分,或者也可以scan你指定的键长(同前面),同时也优化其它的结构。不难看出,这个关键词就是为了帮助我们获得合适的初猜结构。举个例子,假如我们现在确定了反应发生的位点,它可能是A原子与B原子的成键,那么我们就可以在建模的时候将这两个模型的距离放的稍微远一点(比如增大0.4埃到0.5埃),然后我们就可以用modredundant固定这个键,这样这个键长就不会发生变化,而对其它结构进行优化,这样一方面我们可以获得较为合理的其它部分的结构,而被我们固定的这一个键也会因为力的变化,在下一步进行过渡态计算时而优先被选为计算的位点。
因为我这是直接参考的文献,文献中直接给出了过渡态的结构信息,所以我就可以直接照着它的信息建模一个差不多的结构,这里其实也是耽误了很长的时间,我发现这里由于EC的环会被打开,所以在获得初猜结构的时候不能只考虑Na离子与两个O之间的距离,还需要调整一下将要断开的C-O键之间的距离,我最开始就是仅调整了Na离子与两个O之间的距离,但是发现不能获得过渡态结构,后面好不容易找到一个只有一个虚频的结构,但它的频率又太小了,不能算作是过渡态,跑IRC也没能得到初始结构以及产物。以下是我初猜的结构图:
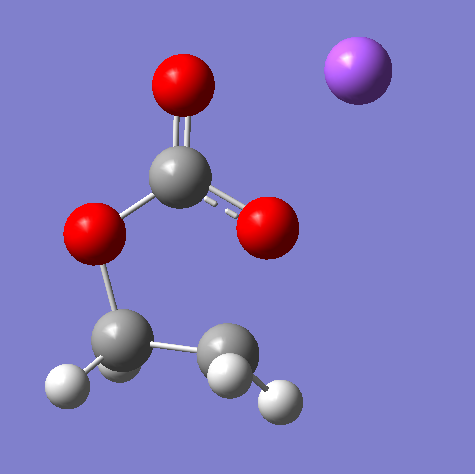
其实就是除了调整Na与氧的距离之外,还调整了需要断开的键长,这样键长偏长之后重新打开这个gjf文件就不会显示成键了。我直接用的这个结果进行的下一步过渡态搜索,其实如果对自己猜想的结构不放心的话可以在这里再做一步柔性优化,我这里就是人家文献里面都直接给出了这个模型,已经告诉了我这个模型就是过渡态结构了,所以我不需要做柔性优化,要是自己做自己的体系我想还是需要在这里做一步柔性优化的,避免出问题,这里将我之前尝试柔性优化的gjf文件贴出来:
1 | |
其中前面的opt=modredundant就代表启用柔性优化,后面的B则代表bond,数字为成键的信息,比如10号原子和11号原子之间的键,F则代表fix,固定,如果是scan的话则为s,后面需要跟上两个数字,分别为扫描多少步以及每一步的步长。
过渡态搜索
其实有了合理的初猜结构,后续找到过渡态就是水到渠成的事儿了,我将我寻找过渡态的gjf也贴出来吧:
1 | |
我发现我这里在计算Hessian矩阵时如果设置为calcfc的话很容易就报错不收敛,所以我就直接用calcall来高精度计算,时间上反而并没有增加多少,因为使用calcfc在报错之前往往还会运行很长的时间(震荡),而这个时候calcall早就已经收敛了。
过渡态计算结果如下,我把频率也一并贴出来了,计算出来的结构的各项参数也与文献中的结果基本一致:
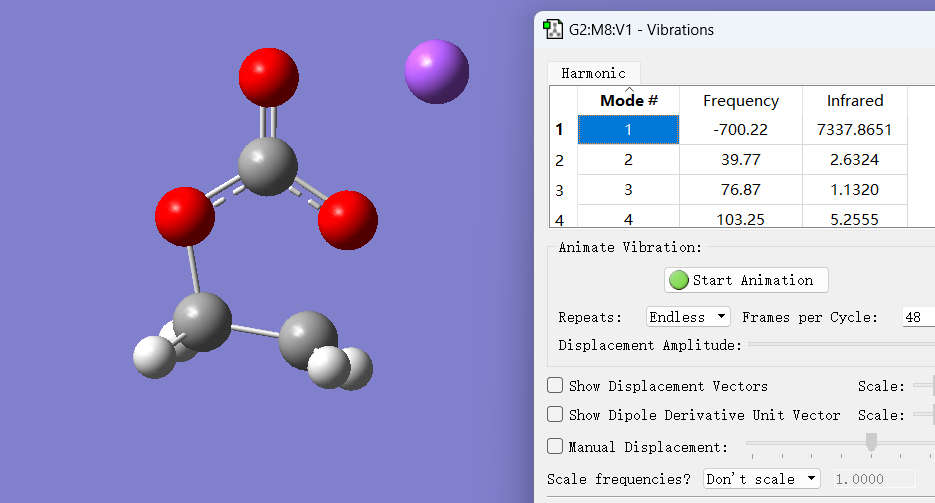
我认为一个比较合理的过渡态首要满足的条件就是它的虚频要足够大,然后就是它这个虚频振动的方向是你所期待的发生反应的方向,当然最重要的还是下一步的IRC验证能够满足反应物和产物的倾向。
IRC验证过渡态是否合理
IRC计算可以参考帖子:
在Gaussian中计算IRC的方法和常见问题 - 量子化学 (Quantum Chemistry) - 计算化学公社 (keinsci.com)
对于我们计算得到的过渡态,它其实就是势能面上的一个鞍点,这个鞍点可能是我们所期望的反应物与产物之间的鞍点,但是也有可能是其它反应的鞍点,所以IRC就是从你得到的过渡态结构出发,向鞍点的两边进行运算,不断的降低能量,这样计算出来,只要运行的步数够长,那么我们就可以获得这个过渡态它对应的反应物以及产物。
使用IRC的时候也很简单,以下是我计算IRC的时候用到的关键词:
1 | |
calcall就不用多说了,每一步都重新计算Hessian矩阵,牺牲计算时间来换取更高的精度并避免报错,lqa就是表示采用LQA方法来跑IRC,Gaussian内置了三种方法来进行IRC计算,具体可以参看上面的帖子以及官方手册,不过简而言之就是LQA是最容易收敛的方法,maxpoints就是指定跑多少步,stepsize则是指定跑的步长,这两个可以根据自己的需求设置,很好理解,reverse这个关键词是选填的,我们假设反应物到产物是一个方向,那么过渡态在中间,这个时候要跑IRC那么它其实是有两个方向可以走的,一个是向产物的方向,一个是向反应物的方向,所以这里的reverse其实就是与forward关键词对应,表示跑IRC两个不同的方向,当然也可以不设置这个关键词,那么Gaussian就会把两个方向都跑一遍。注意,Gaussian是不知道哪个方向是反应物,哪个方向是产物的,forward和reverse只是一种表示罢了,具体是产物还是反应物还是需要我们自己根据跑出来的结构进行判断。这里其实设置forward和revers分开跑的原因其实就是为了放置报错,这样如果forward没有报错,但是reverse报错了也方便修改查找原因。
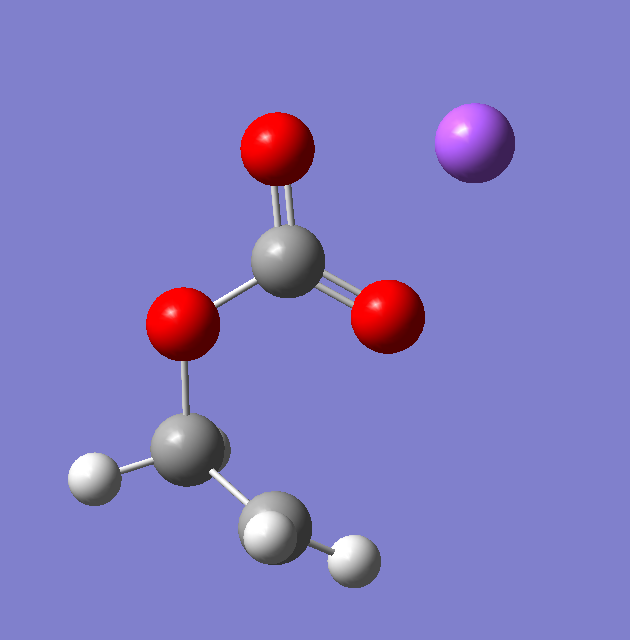
这里将我计算得到的Forward与Reverse的结构贴出来:
以下是Forward关键词跑了50步之后的结构,与文献中过渡态的下一步结构非常类似:
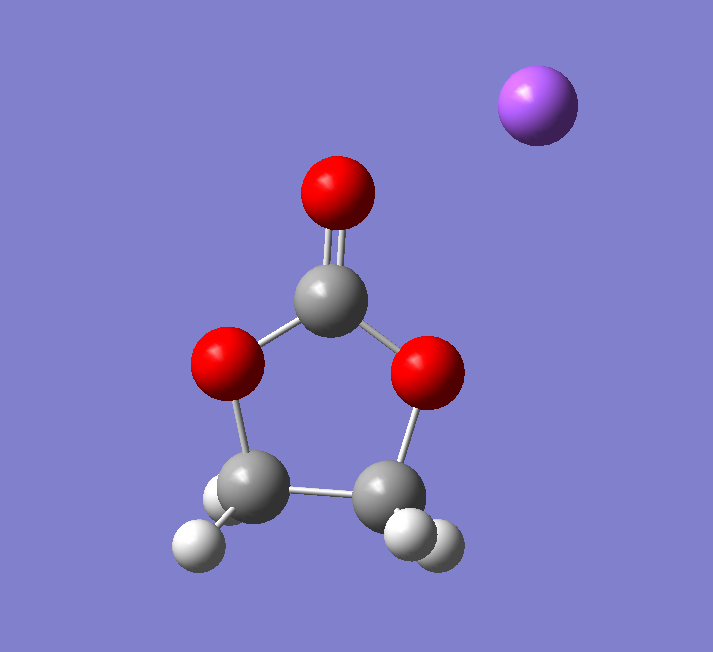
以下是Reverse关键词跑了50步之后的结构,与我们寻找过渡态之间得到一个电子的结构非常类似:
注意,想要通过IRC来直接获得产物和反应物是比较困难的,除非你设定一个足够长的步数,所以下一步我想应该是需要根据IRC获得的产物来进一步结构优化,才能获得真正的产物结构。
不过从上面的计算结果来看,这个过渡态就算是找对了,无论是从文献上的验证还是所自己计算的角度进行验证,都是没有什么问题的。
总结
Gaussian计算过渡态其实也没有想象中的那么难,用ts关键词来计算更能帮助我构建这种不知道产物的模型,此外还有ts2等关键词以后也可以根据需求进一步尝试,总结一下使用ts关键词进行过渡态计算的步骤吧:
- 建模并进行反应物的结构优化,获得合理的反应物构型。
- 获得过渡态的初猜,具体可以根据自己的化学直觉建立模型,然后采用柔性优化获得更加准确的模型。
- 使用ts等关键词进行过渡态搜索,对结果查看虚频,并自己判断是否达到需求。
- 使用IRC进行验证,看自己计算得到的过渡态是不是自己所期望的反应的过渡态。
过渡态搜索其实说难不难,说简单不简单,这种东西更需要的或许是经验,有经验的话做起来就会相当的快,没经验的话可能兜兜转转还会得到一个错误的结果,多做多总结才能让我们下一次搜索过渡态是做得更好更快。