Gromacs的学习以及应用(五)
接下来继续复现之前讲的文献中的内容,先看看正文里面作者算了哪些性质:
其实看上去还是很简单的,最开始先有一个ESP的计算,这个是通过量化计算方法得到的,我在前面有过简单的描述,然后就是径向分布函数(Rdf)以及由它计算得到的配位数(Coordination Number),最后就是统计了不同体系的氢键数量。
表面静电势ESP(Electrostatic Potential Surfaces)的计算
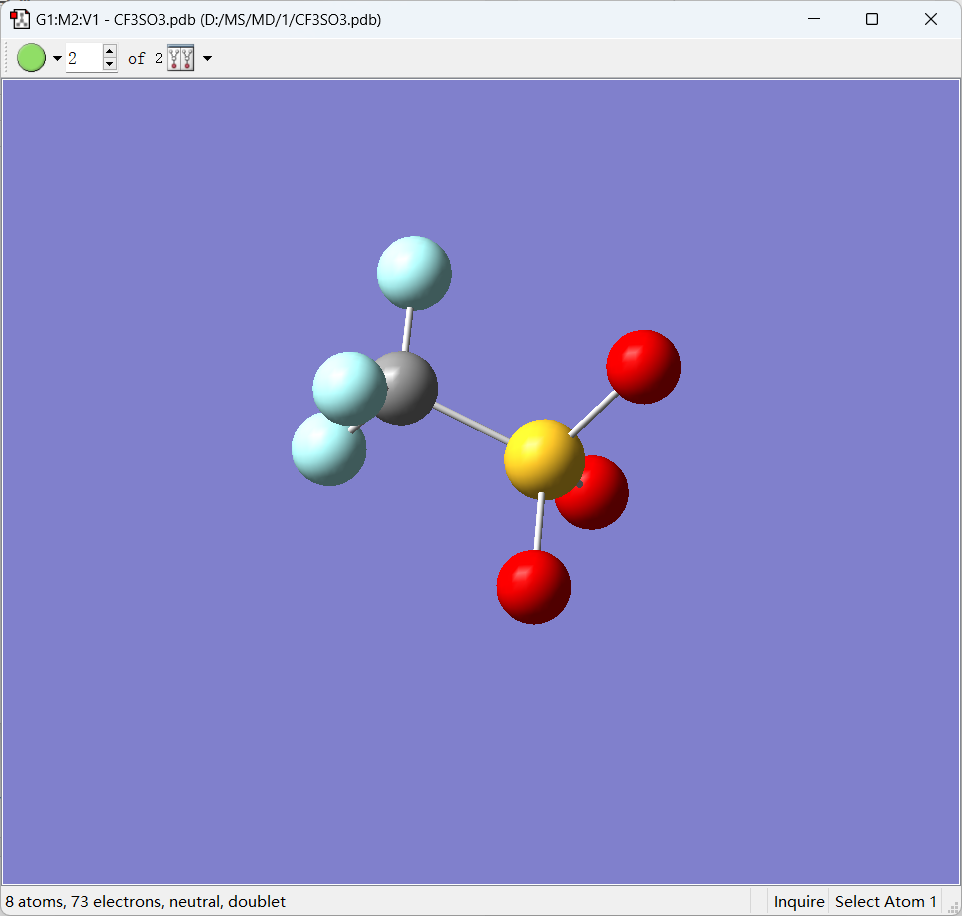
通过Gaussian来计算分子的ESP,这里以CF3SO3为例,可以直接用之前建好的pdb模型导入到GaussView中。
直接保存为CF3SO3.gjf格式的文件,其它模型也可以直接在GaussView中建模。
然后将所有的gif文件上传到服务器上,依次修改文件的开头并删除文件结尾的成键信息,这里我计算用的关键词为:
1 #6-311++G(d,p) em=gd3 b3lyp opt scrf=solvent=water freq
这也是参考文件写出来的,然后用formchk将计算得到的chk文件转化为fchk文件,再将其导出到电脑上并用GaussView打开。
这里可以参考这篇文章,有详细讲述如何通过GaussView来绘制ESP图:
blog.molcalx.com.cn/2018/12/19/gaussian-esp.html
我根据它的步骤然后自己修改了一下显示的范围,即可以获得以下的图,注意,GaussView中的单位为au,对于能量来说也就是Hartree,那么上面的范围其实想要转化为eV的话是需要乘以一个27.2114的,这样相乘之后的结果与文献中的基本无异,但是这个颜色跟文献中是完全相反的,需要注意一下:
其它分子的计算结果其实都是差不多的,这里就不全部放出来了,那么对于ESP的计算结果也就基本上获得了。
RESP电荷(Restrained ElectroStatic Potential )的计算
在上一篇博客中,对于CF3SO3阴离子,我们需要去计算它每个原子上的电荷分布情况,因为文献中直接给出了,我们可以直接参考,但是对于NO3以及SO4阴离子,我们需要通过计算RESP电荷来确定不同原子上的电荷,以便于我们进行后续的MD模拟。对于这部分的计算其实我在最开始的博客中有过简单的介绍,参考
RESP拟合静电势电荷的原理以及在Multiwfn中的计算 - 思想家公社的门口:量子化学·分子模拟·二次元 (sobereva.com)
这篇博客基本上就能很容易的将小分子的电荷信息获取到。接下来我以NO3为例演示一下。
根据上面计算ESP的步骤,这个时候我们已经有了NO3结构优化后的chk文件,并用formchk将其转化成了可以供其它软件读取的fchk文件,然后启动Multiwfn,输入NO3.fchk导入文件,接下来依次按照顺序输入:
1 2 3 4 7 //布居分析与原子电荷计算
按照上述操作后目录下就会多出来一个NO3.chg的文件,有5列内容,分别为原子名,x,y,z,电荷。然后可以根据这个信息将其整合到MD模拟的topol文件中。
NO3以及SO4的力场参数的获取
根据参考文件,NO3以及SO4力场文件的获取是通过ACPYPE来获取的,这里我采用了更为便捷的方法来获取它们的力场参数,那就是采用sobtop这个软件,具体可以参考sobtop的官网:
Sobtop (sobereva.com)
这个软件目前来看,要用的话必须在它所在的目录,并且将需要生成top文件的mol2文件也copy到它的目录下才行,然后先要指定一下原子类型,再根据Amber力场以及GAFF力场生成top文件,此外它还可以根据计算得到的chg文件直接将电荷信息整合到top文件中。以下是我使用sobtop生成的NO3.top以及NO3.itp文件:
NO3.top文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 ; Created by Sobtop (http://sobereva.com/soft/sobtop) Version 1.0(dev3.1) on 2023-02-25
NO3.itp文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 ; Created by Sobtop (http://sobereva.com/soft/sobtop) Version 1.0(dev3.1) on 2023-02-25
然后参考之前计算Zn(CF3SO3)2的计算步骤,整合一下计算Zn(NO3)2所需要的top文件以及itp文件,对于Zn离子以及水的itp文件可以直接借用上次计算的文件,然后需要修改一下nonbonded.itp文件以及top文件,主要是将之前的CF3SO3的信息删除再加上此次NO3的信息,依然是模拟36个Zn盐,因此需要72歌NO3离子,以下是修改后的toppl.top文件:
1 2 3 4 5 6 7 8 9 10 11 12 #include "nonbonded.itp"
然后是重新整合一下nonbonded.itp文件,如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 [ defaults ]
最后便是将生成的NO3.itp文件最开始的[ atomtypes ]的信息去掉即可,这样结合上次的计算准备,我们就有了topol.top,water.itp,Zn.itp,NO3.itp,top文件也就基本准备好了。
对Zn(NO3)2、ZnSO4以及Zn(Cl)2的分子动力学模拟
结合我之前对Zn(CF3SO3)2的练手计算,我们现在就可以获得计算所需的top、itp文件以及它们各自的pdb文件,这样我们接下来就可以直接采用之前提到的packmol软件生成一个包含有1000个水分子以及36个Zn盐的盒子,再转化成gro格式的文件,就可以开始计算了,采用的mdp文件与之前一致,依然是五步计算。
后续继续完成对SO4以及Cl离子的计算,不得不说这些操作步骤真的是相当的繁琐且无趣,大量的工作都是重复性的,而且各个软件之间来回倒腾,真的是相当的折磨人了,这里先挖个坑,以后想办法写个脚本出来,能够直接从结构文件到提交任务,缩短整个工作流,这里我在计算Zn(Cl)2时实在受不了了,简单写了一个脚本,可以自动持续的提交任务,一次性就可以将五个步骤依次计算完成(其实现在看来分为5步计算会不会有点过于冗余了,可能以后还需要精简)。以下为我写的代码,为了让文件看上去更加有序,所以我还是将不同步骤的计算安排在不同的目录中,这样产出的文件也能到相应的文件:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 #!/bin/bash
对分子动力学模拟计算结果提取Rdf并通过python脚本求配位数
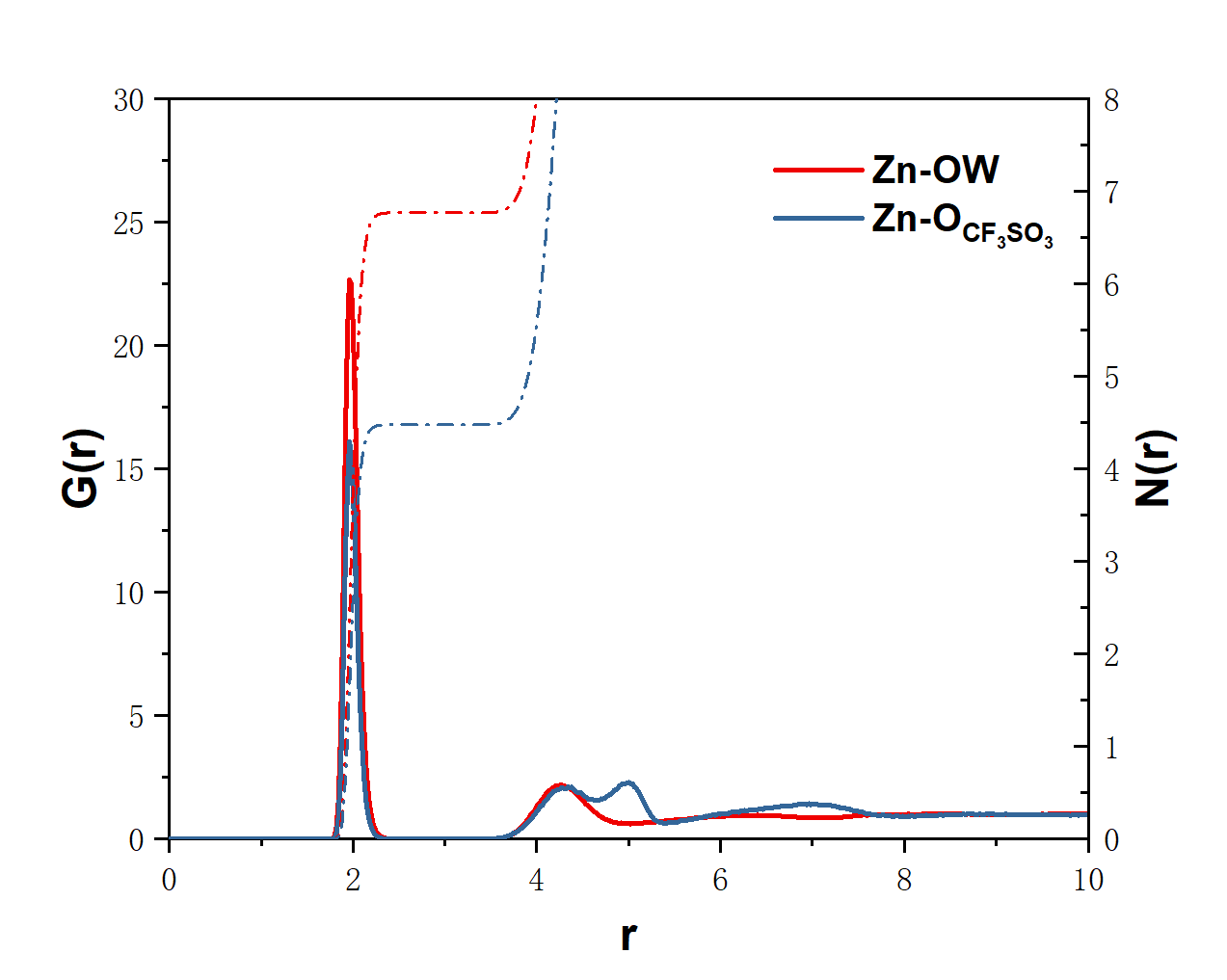
关于提取Rdf的方法在前面我已经讲过,这里为了方便与文献中 的结果,就再次以Zn(CF3SO3)2为例,演示一下,首先创建一个索引文件,文献中对比了Zn与阴离子以及水分子中的径向分布,所以我们就按照这个来创建索引文件:
1 2 3 4 5 gmx make_ndx -f prd.gro
这里OW就表示溶剂水中的氧原子,O则表示阴离子中的氧原子,这里就会输出一个index.ndx文件,然后就可以继续提取以Zn未参照的关于这两种氧原子的Rdf信息,输入:
1 gmx rdf -f prd.xtc -n index.ndx
然后根据提示,先选择Zn离子作为参照,再依次选择OW以及O的组来输出rdf信息。
输出后回得到一个rdf.xvg文件,将其转移到自己的电脑上,我先删去了文件开头的一些注释内容,只保留数据信息,然后改后缀为dat,将其导入到origin中进行作图,因为要用到积分等操作,所以在自动化的脚本写出来之前,还是得用origin来手动操作。
对于rdf的绘图非常简单,给出的数据直接按照 r − g ( r ) r-g(r) r − g ( r ) N ( r ) N(r) N ( r )
N ( r ) = 4 π ρ ∫ 0 r g ( r ) r 2 d r N(r)=4{\pi}{\rho}\int^r_0g(r)r^2dr
N ( r ) = 4 π ρ ∫ 0 r g ( r ) r 2 d r
这个计算还是很简单的,这里说一下数密度ρ \rho ρ ρ \rho ρ
ρ = N V = ( 1000 + 36 + 72 ) 3 2 3 = 0.033813 \rho=\frac NV=\frac {(1000+36+72)}{32^3}=0.033813
ρ = V N = 3 2 3 ( 1000 + 36 + 72 ) = 0.033813
那么在origin中先设置一列的列值等于g ( r ) ∗ r 2 g(r)*r^2 g ( r ) ∗ r 2 4 π ρ 4\pi\rho 4 π ρ
我觉得这里有可能是我对Zn原子的一些基本信息没有定义对,或者是计算所用的方法跟文献中有差异,此外我想模拟的时间应该也会造成影响,这个就留待以后再进行探究了吧。研究了一下,还是写了一个简单的python脚本来直接获取我们的绘图需要的数据,以下是实现的代码,使用这个代码之前要记得对xvg的文件使用sed进行清洗,将文件中以@开头的行转化为#开头的内容。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 import numpy as np'rdf.xvg' , comments='#' )0 ] * 10 1 ]0.0338134765625 for i in range (1 , len (r)):4 * np.pi * rho * np.trapz(gr[:i] * r[:i]**2 , r[:i])'output.dat' , np.column_stack((r, gr, n)), fmt='%.6f' )
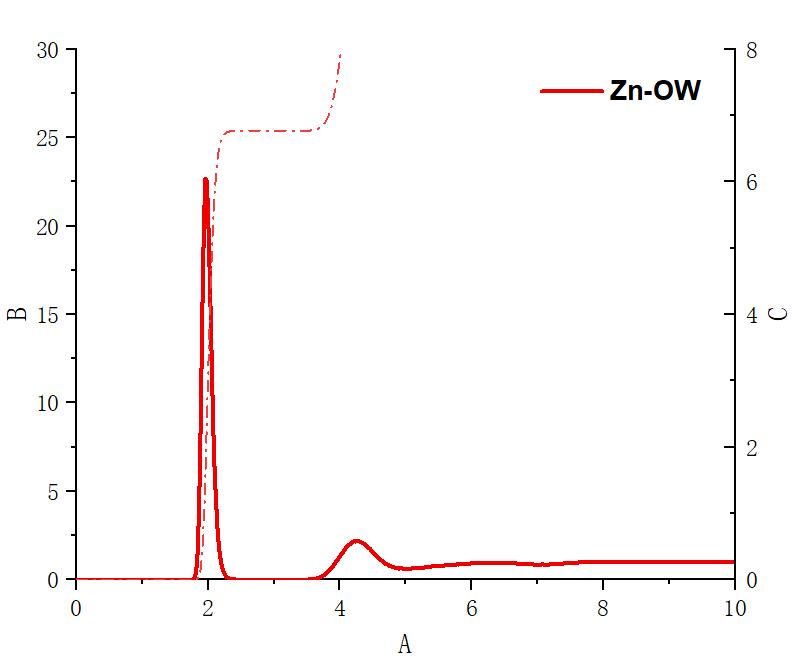
这个脚本目前可以实现一组数据的转化,更多组的数据转化以后再研究了,以下是根据它的转化结果绘制的上面已经绘制的图,没有啥错误的:
因为懒惰,所以坐标轴还有一些画图的细节我就没有调整了,结果还是没啥问题的,具体写脚本的细节这里就不多赘述了。
对H-bonds数量的提取
对这个数量的提取相当的简单,文献中探究的是water cluster的H-bonds 数量,我对此的理解就是模拟中水分子之间的氢键,所以首先在界面中输入:
1 2 gmx hbond -f prd.xtc -n index.ndx -s prd.tpr
不过这里其实可以在-f时输入gro文件,这样就只会统计gro文件那一帧的氢键,其实跟统计平均值是差不了太多的,我这里计算出来1000个水分子大概有1450个左右的氢键,平均算下来每个是1.45的样子,跟文献中也算是有较大的偏差了,不过这里我突然想到会不会是盒子大小的原因,因为文献的SI里面并没有指出模拟所用的盒子大小,我觉得它用的盒子可能会比我的这个盒子还要小一些。
总结
对Gromacs的学习基本上就算是完成了,至少是掌握了基本的使用方法,以后我的计算模拟的尺度选择面就更广阔了,不过还是有许多的东西是需要我慢慢探究的,想要真正的完全学会这个软件还是任重而道远啊,继续努力加油吧。