Gromacs的学习以及应用(四)
Gromacs的学习以及应用(四)
这里就不贴复现的文章内容了,有兴趣的可以去看前面的博客
简介
上一章中解决的其实是Gromacs的外源性内容,即无论是top文件还是结构文件,这些都是要我们通过自己的手段去获得的,哪怕是Gromacs自带的力场参数,也是要我们自己进一步改造编写,从而生成自己的top文件,而结构文件则需要我们自己去建模,然后还要用Gaussian这样的量化计算软件去先帮我们把小分子结构优化了才行。现在这一部分内容就是对Gromacs内源的一些操作,如编写mdp文件,生成tpr文件并提交任务开始计算。
对生成gro文件的补充
之前我都是用的gmx insert-molecules指令来生成盒子并加入我需要的分子结构,但是我发现这个东西在分子数量过多的时候并不好用,就拿前一章的1000个水分子+36个Zn(CF3SO3)2举例,如果你是先在盒子中加入72个CF3SO3就还可以将1000个水分子加进去,但是先添加1000个水分子再添加CF3SO3就不行了,所以我这里想试试用packmol来生成盒子。
对于packmol的安装和使用可以参考这篇文章:
分子动力学初始结构构建程序Packmol的使用 - 思想家公社的门口:量子化学·分子模拟·二次元 (sobereva.com)
使用packmol就需要在目录下编写一个.inp文件作为它的输入文件,就是对它生成盒子做一些设定,我的.inp文件如下:
1 | |
Packmol里面的单位都是埃,所以这里我们要设定一个3.2nm的盒子大小,输入文件里面对应的就是32,tolerance [数值]就是要求原子间距离不能小于多少,一般设为2.0。add_box_sides 1.2,则会给盒子各方向再增加1.2埃,避免做模拟时有某些边界的原子和镜像盒子里的原子间存在不合理接触,output box.pdb则指定输出文件保存为box.pdb,至于Structure部分就很通俗易懂了,包括输入文件,生成的个数,以及在多大的盒子内生成,最后以end structure结束。
将这个文件保存为mix.inp文件,再直接输入:
1 | |
packmol就会运行并计算比较合理的初始结构:
得到了box.pdb文件之后就可以通过gromacs自带的gmx editconf指定将其转换为gro文件:
1 | |
以下就是输出结果:
mdp文件的编写
对于mdp文件的编写其实我现在也没有一个很好的思路去讲解,我现在用的mdp文件其实是参考的网上教程,可以参考这个网站的内容来思考自己的mdp文件该怎么编写:
这里他给出了5个mdp文件,先是两次能量最小化的计算,然后是两次预平衡的计算,最后一次成品计算。这里我参考文件内容以及上面网站的说明给出自己的解释,以下是对第一个mdp文件也就是min.mdp文件的一些注释:
1 | |
第一次能量最小化的目的是调整分子的初始位置, 这样使用SETTLE约束算法时就不容易遇到错误了,第二次能量最小化就是为了采用SETTLE算法进一步计算,这时就可以将define = -DFLEXIBLE删去。
接下来再来看看第一次预平衡的mdp文件:
1 | |
第一次预平衡也就是所谓的NVT平衡,这里给出的解释是:使得体系在加压耦合之前达到正确的温度(298.15 K). 因为如果同时添加温度和压力耦合可能会导致系统不稳定并发生崩溃. 但是我在另外一些论坛上看到的解释说,貌似这样分两步运算进行预平衡是没有必要的。接下来看看第二次预平衡的mdp文件添加的内容:
1 | |
第二次预平衡就是添加了压力耦合,此外体系添加的影响更多,运行时间也相应需要更多,所以这里在步长不变的情况下需要提高运行的步数来增大模拟的时长。
最后的成品模拟的mdp文件其实与第二次预平衡的mdp文件类似,我也就放在这里可以看看:
1 | |
成品模拟的主要就是修改了输出信息设置和运行的时长,以及输出信息的密度。
综上,我就完成对mdp文件的解读,对于编写自己需要的mdp文件也有了一个思路。
生成tpr文件
根据前面的步骤,现在理论上来说我们已经有了topl.top、conf.gro以及min.mdp文件,那么我们这一步就可以直接用Gromacs自带的gmx grompp指令来生成tpr文件,直接输入以下的指令:
1 | |
这里因为我们对gro文件和top文件都是按照默认名称来命名的,所以这里就无需通过-c和-p选项来指定输入的gro文件以及top文件,gromacs会自动将当前目录下将conf.gro和topl.top文件输入,-f命令就指定了输入的mdp文件,这里是第一次能量最小化的min.mdp文件,-pp和-po就是输出经过gromacs处理后的mdp文件以及top文件,其中前者会将gromacs所有可以设置的参数罗列出来,没有设置的就会按照默认的值输出,如果自己在mdp文件中进行了设定的参数则会将其改为设定的参数。后者则是将所有引用的itp文件整合进top文件中,打包成为一个文件。以下为成功运行的界面:
运行Gromacs并提取计算结果
提交计算任务
获取了tpr文件之后,我们就可以提交任务,开始模拟计算了,可以参考我之前写的运行Gromacs的任务指令,这里我将脚本命名为gromacs.sh,注意需要将脚本中的最后的eql改成自己的mdp文件的名字,比如这里就应该改成min
1 | |
保存gromacs.sh后在命令行中输入:
1 | |
就可以运行任务了。
运行结束后目录下会多出来许多文件,包括min.edr, min.gro, min.log, min.trr以及任务管理系统的.out文件,文件所包含的信息我在之前已经讲过,这里就不多赘述。接下来可以直接开始第二次的能量最小化计算,将上一步计算得到的min.gro文件以及提交任务的gromacs.sh复制到新的文件夹2min中,再将已有的min2.mdp文件复制到相同的文件夹中,同时将上一步gmx grompp生成tpr文件时同时输出的min.top文件也复制到2min文件中,这样2min文件中将会有min2.mdp, min.gro, min.top以及gromacs.sh四个文件,那么可以在这个目录下输入:
1 | |
后续就是继续将gromacs.sh中的文件名改一下,改为min2,然后提交任务,以此类推,我们可以很快的完成后续的两次预平衡以及成品模拟。
在第二次预平衡时,生成tpr文件时产生了Warning,如下
With Nose-Hoover T-coupling and Parrinello-Rahman p-coupling, tau-p (2) should be at least twice as large as tau-t (2) to avoid resonances.
为了消除这个Warning,我没有使用-maxwarn 1这个指令,而实将tau-p的值设置成为了4,这样或许会增大一些计算量,但是可以避免Warning。当然,在最后的成品模拟中我也做了类似的修改。
这里成品模拟计算所花费的时间比之前四个步骤加起来的时间还要多。
提取计算结果
先查看一下能量最小化的结果,在第一次能量最小化的目录下输入:
1 | |
然后选8 Potential,再连续按两次Enter键,就会将能量信息输出到min-energy.xvg文件中,类似的可以在min2目录下也这样输出min2-energy.xvg,为了便于后续采用gnuplot这个软件绘图,可以将文件中的@字符全部替换为#,使用以下的bash命令:
1 | |
其中,-i选项表示直接在原文件中进行修改(in-place),s命令表示进行替换操作,g选项表示替换所有匹配项,不仅限于第一次出现的位置。
随后用gnuplot绘图,这里我把我绘制的plt文件也贴出来吧,这个是在chatGPT的帮助下写成的:
1 | |
这样的画出来的图如下:
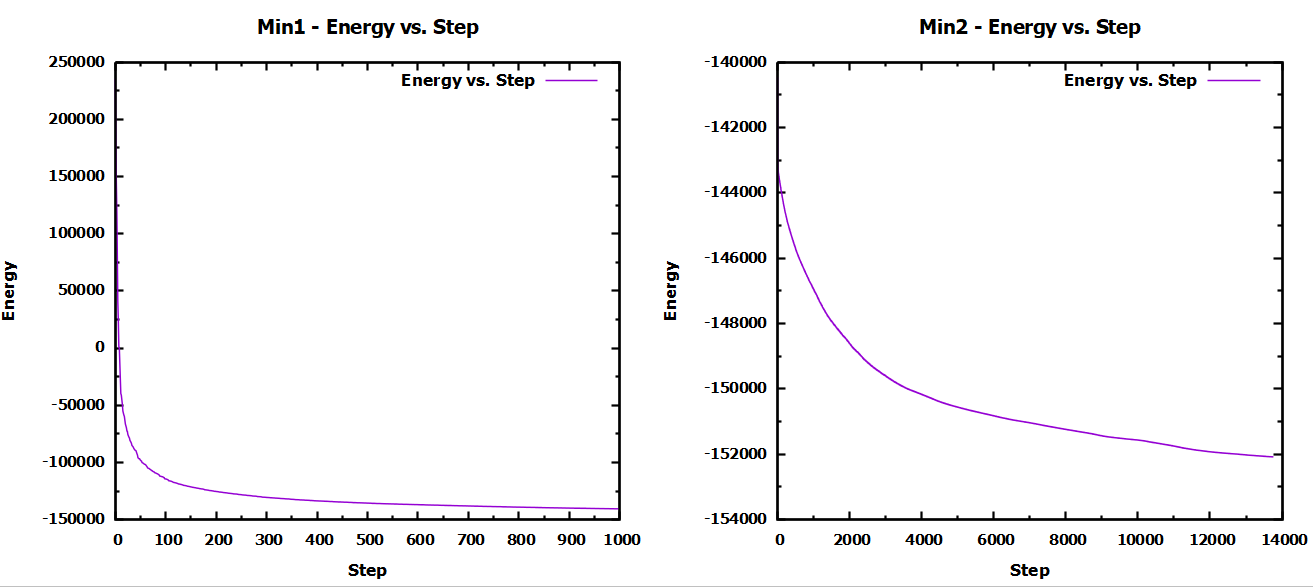
感觉我这里第二次能量最小化可能时间还不太够,正常或许应该时要能量基本没有变化了才最好。
接下来是验证NVP和NPT预平衡中温度和压力的耦合情况,所以我们需要导出模拟时间与温度、压力的关系,类似于上面导出能量的操作,我们在eql和eql2的目录下输入
1 | |
NVT也就是第一次预平衡需要选择12 Temperature,NPT第二次预平衡则选择14 Pressure。
导出数据后再用上面的sed指令对文件进行清洗,再讲上面的plt文件拿来改一下,就可以得到下面的图片:
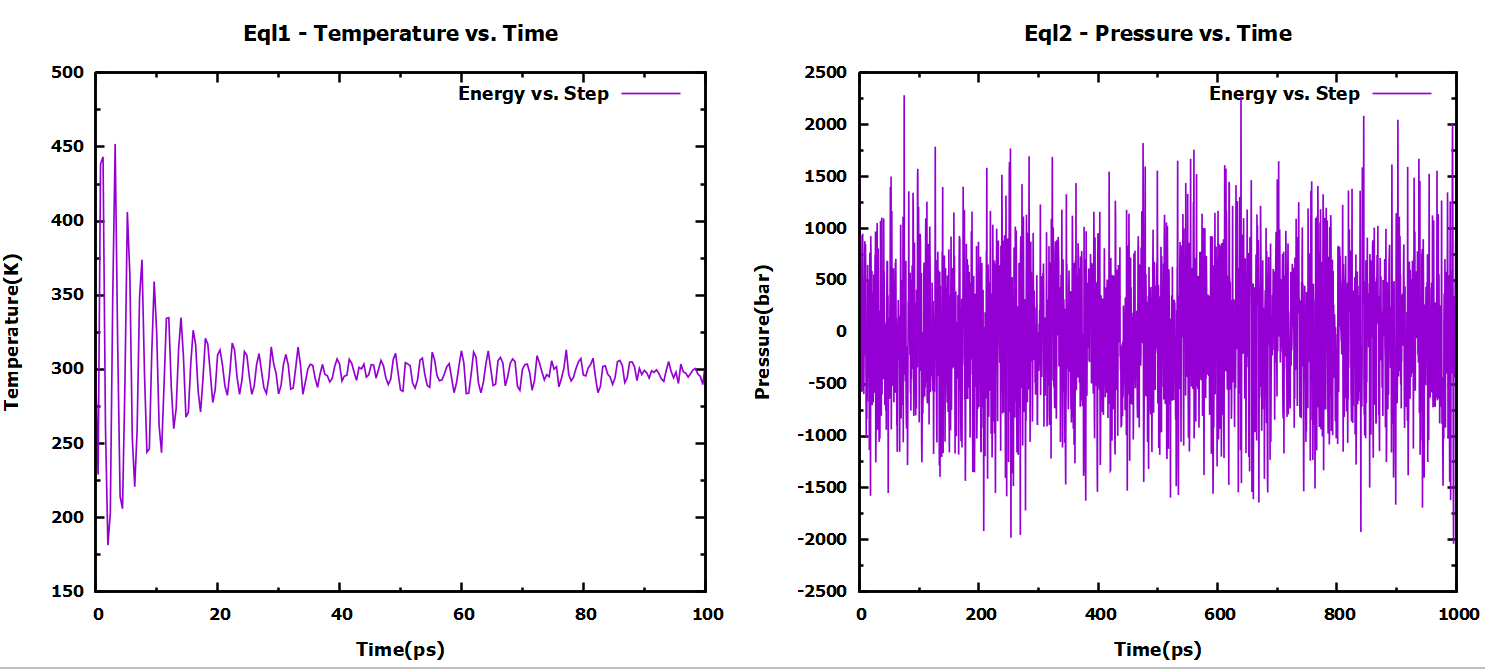
这里的压力波动很大,但是只要平均值接近1bar即可
接下来是成品模拟的结果提取,我们可以先导出一个轨迹文件来看,毕竟看着分子振动还是很酷的,首先需要在成品模拟的目录下输入
1 | |
这是为了防止后续在VMD中查看轨迹时存在贯穿整个模型的键。
导入VMD中的时候,在VMD这个软件里面用cd切换工作目录到保存prd.gro和prd-mol.xtc的目录下,输入以下代码
1 | |
就可以看到分子结构及其变化了,以下是我这次的计算的一个快照:

接下来尝试一下导出rdf也就是径向分布函数,这里需要创建一个索引文件,对于索引文件我目前的理解是创建一个组,方便我们对它进行后续的操作,比如正常情况下我们创建的gro文件会将其按照离子,分子以及溶剂等自动分成好多个组,但是当我们想做两个原子之间的rdf时这种分组方式就不够用了,所以我们需要自己按照自己的需求创建组,一遍gromacs处理数据,使用以下指令来创建组:
1 | |
这里我就按照原子新建立了四个组,目录下会生成一个默认名称的index.ndx文件。比如接下来我想要探究Zn离子附近的分子分布情况,那么我就可以根据上面我创建的组进行数据分析,使用gromacs的rdf指令:
1 | |
然后选择以Zn为参考组,然后依次选择其它想要分析的元素,选择好之后按Ctrl+D即可输出rdf.xvg文件。
记得也用sed清洗一下,然后就可以用gnuplot作图,如下:
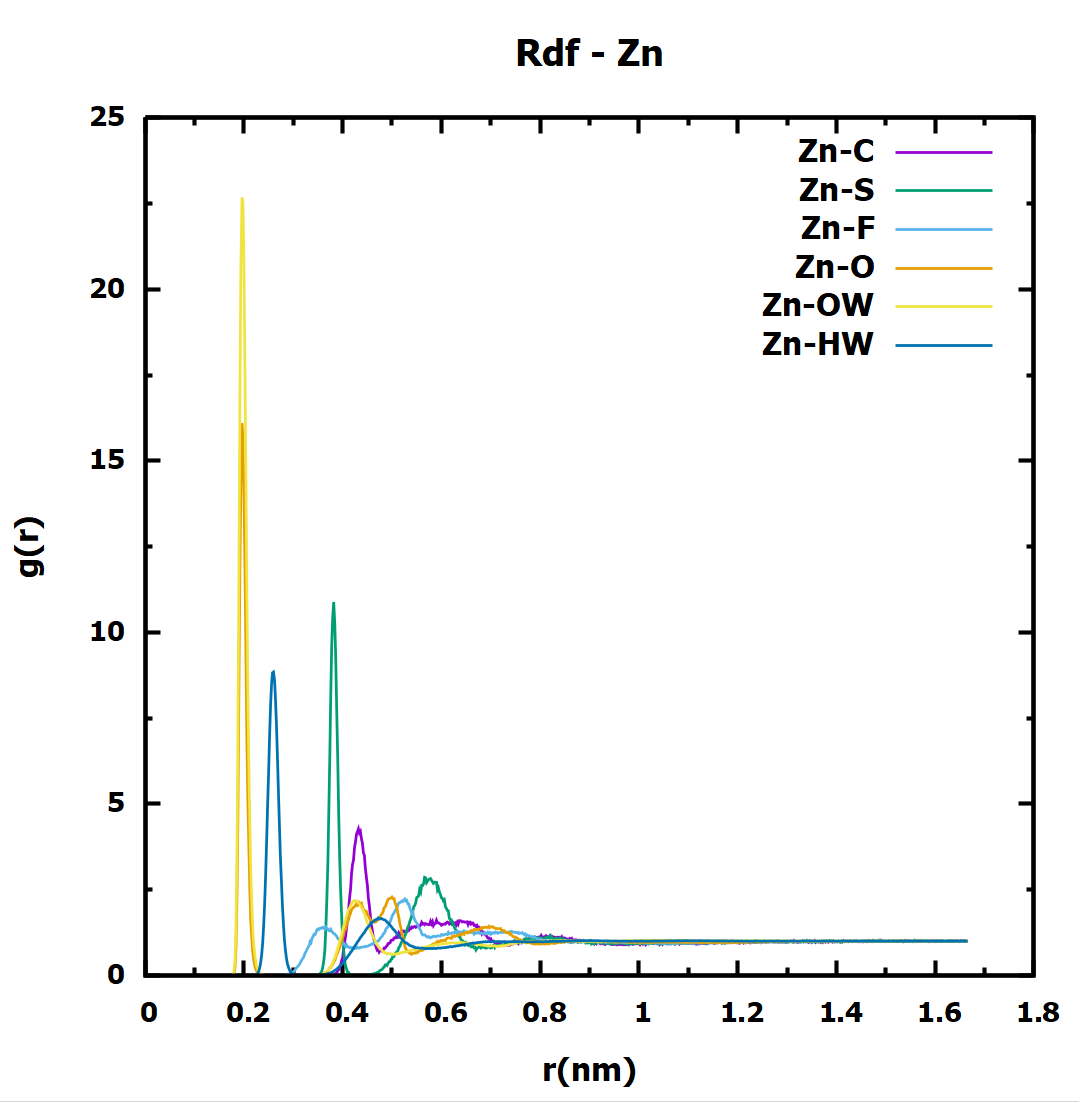
可以看出来溶剂化效应还是比较明显的,Zn离子基本上完全被水分子包围。
小结
综上,对于Gromacs的练手基本就完成了,整个工作流就确定了,不过注意,我这里的工作流应该是仅适用于小分子的材料体系,如果是处理蛋白质或者其它生物大分子,那么这个工作流可能就没有那么适用了,接下来可以正式着手复现文献内呈现的内容了。