Gromacs的学习以及应用(三)
这里继续对前面一篇博客的内容,先把要复现的文章内容贴在这里:
简介
首先需要尝试的是gmx insert-molecular这个指令,我想要创建一个基于OPC水模型的盒子,包含1000个水分子以及30个Zn盐,这里以Zn(CF3SO3)2为例。
这里插入离子最好的方式我认为应该是genion这个指令,但是genion对输入文件要求tpr格式的文件,我着实很难想到如何在构建模型这一步就将tpr文件输出,所以这里就用请教别人所了解到的insert-molecular方法。
Top文件的编写
看文献内容可以看出,使用的是Amber03力场,而Gromacs是自带有Amber03力场的,所以我们可以先把Amer03力场的参数文件复制到自己的目录下打开,这个参数文件一般在Gromacs的安装目录里面,可以仔细找一下。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 ********************************************************************
这个就是Amber03力场,这里可以看到它还引用了ffnonbonded.itp,ffbonded.itp,gbsa.itp这三个参数文件。所以我们真正要的数据其实是在这三个文件中的。接下来我们需要梳理一下我们需要的参数信息,首先是水模型的信息,但是其实已经不需要在这里获得了,因为前文已经找到了OPC水模型的拓扑文件了,接下来是Zn原子的力场参数,这个的确是需要在这里获得,最后是CF3SO3的力场参数,文献中也指出了,它是根据文献的信息来获取的。
这里先看了一下那三个文件,里面有关Zn的信息只有一行内容,就是在ffnonbonded.itp文件中的(这里我将最上面的atomtype也复制了过来),然后因为Zn元素在我的模拟体系中是离子的形式存在的,为+2价,所以这里的电荷需要修改,此外为了后面能够使用Zn离子的力场信息,我们还需要在后面添加一个[ moleculetype ]以及[ atoms ]信息,因为Zn离子并没有成键信息,所以后面其它的Bonds以及Angles什么的就不用写了,写出来的Zn.itp文件如下:
1 2 3 4 5 6 7 8 9 10 11 12 [ atomtypes ]
接下来是编写CF3SO3的top文件,先看看那篇文献:
Hydrogen bonding in a mixture of protic ionic liquids: a molecular dynamics simulation study - Physical Chemistry Chemical Physics (RSC Publishing)
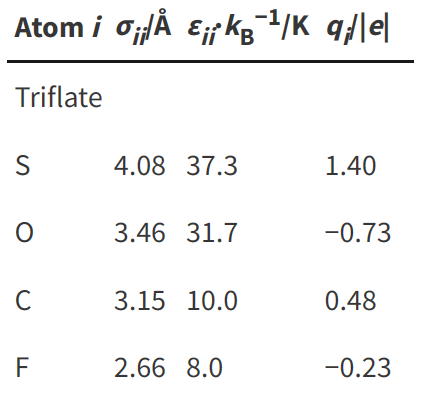
这篇文章给了四个表,罗列出来了CF3SO3的各项信息,我们需要的就是将其编写称itp文件,首先是非成键力场信息,以下是文献的截图:
对应编写itp文件中的[ atomtypes ]信息,这里有几种元素就只写哪几种元素,注意这里再编写的时候要注意单位的换算,比如这里的sigma的单位用的是埃,而gromacs的默认单位为nm,所以要进行转换,对于后面的epsilon,单位为K而默认的单位为KJ/mol,所以要进行转化(需要除以一个120.272)
如下:
1 2 3 4 5 6 7 [ atomtypes ]
还有就是[ moleculetype ]和[ atoms ]信息,moleculetype其实就是给自己的分子取个名字,代表后面的所有信息都是用于这个分子的,也为了在top文件中能够直接引用分子的各种信息。编写atoms信息时应该是分子结构中有多少个元素就要写多少个,如CF3SO3有3个F原子和3个O原子,那么这些都是要写出来的,并且赋予它们不同的编号(ID),写出的[ moleculetype ]和[ atoms ]信息如下:
1 2 3 4 5 6 7 8 9 10 [ atoms ]
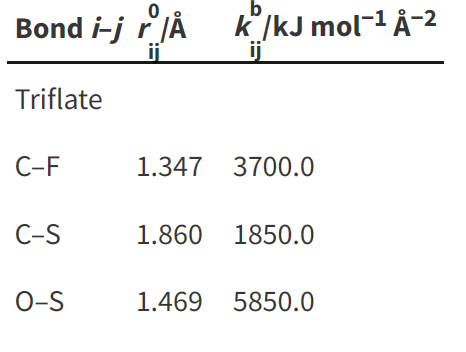
然后是成键信息:
注意在编写这个成键信息的时候是需要将所有原子都给囊括,而且同一个ID不能重复,以下是编写的成键信息:
1 2 3 4 5 6 7 8 9 [ bonds ]
然后就是按照angle以及dihedrals信息继续编写键角和二面角信息:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 [ angles ]
这里我是把所有等价的键角和二面角都罗列了出来,但是听说有可以不把这些信息都写上的办法,所以可能这里还需要进一步学习。
综上,我就将CF3SO3的itp文件写完了,这里我就已经有了三个itp文件,我把它们分别命名为Zn.itp,water.itp以及CF3SO3.tip,但是在这里需要注意的是,对于能够让gromacs处理的top文件,是有严格的格式要求的,第一个关键词应该为[ defaults ]关键词,代表一些基本的参数设置,然后是非成键信息,也就是我们的[ atomtypes ]信息,[ atomtypes ]可以出现多次,但是它必须得是在所有得[ moleculetype ]之前,而[ moleculetype ]之后则是每一个分子的信息,两个[ moleculetype ]之间就代表一个完整的分子信息,在文件的最后才是[ System ]以及[ Molecules ]信息,其中[ System ]信息其实就是给自己的体系取一个名字,可以随便写,而[ Molecules ]则需要根据你的gro文件来编写,比如我先往盒子里面添加了10个水分子,再添加了5个Zn离子,最后再添加了10CF3SO3离子,那么[ Molecules ]中的信息也应该是按照这样的顺序去写。
我们可以看到,所有我们现在编写的itp文件里面都有[ atomtypes ],但是[ atomtypes ]又必须在前面,所以我们可以再编写一个nobonded.itp文件,专门用来存放所有[ atomtypes ]的信息,此外为了精简,我们可以将[ defaults ]的信息也写在这个文件的开头,这样咱们在include这个文件的时候就会自动写入[ defaults ]的信息,在top文件里面我们就不用再写了,我写的文件内容如下:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 [ defaults ]
注意,在写了这个文件之后,之前的三个itp文件里面的[ atomtypes ]信息应该要对应的删去。
然后我们可以开始写top文件了,我们想要模拟的是1000个水分子中溶解了36个Zn盐,也就是36个Zn离子和72个CF3SO3离子,那么写成的top文件如下:
1 2 3 4 5 6 7 8 9 10 11 12 13
这里前面四行就是引用对应的itp文件,注意这四个文件应该与top文件在同一目录下,此外nonbonded.itp应该是严格放在第一个的,其余三个的先后顺序则不重要,[ Molecules ]后面的顺序则应该是与后面我们创建盒子时添加分子的顺序一致,这样我们的top文件就创建完成了,可以将它保存为topl.top。
创建gro文件
gro文件的创建就相对简单了许多,对于Zn离子的创建,我就是直接用MS画了一个Zn原子,导出pdb格式文件,再用gromacs的gmx editconf指令改了一下格式,水模型的文件因为我们是采用的一个四点水模型,所以我这里借用了gromacs自带的四点水模型的gro文件(与之前找力场文件类似),拿过来把多余的水分子去掉,只保留一个水分子的结构信息即可,然后对于CF3SO3阴离子的结构我则是通过GaussView画了一个模型,然后导出pdb格式(这里用GasussView的目的其实是想用Gaussian优化一下的,但是我懒得操作了,直接用画出来的结构了,最好还是应该优化一下才行。),再就是跟Zn离子的操作类似,转化为gro文件即可。这里我将三个文件的信息写在下面:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 # 水模型
注意,上面这三个分子应该是分别保存在三个不同的文件中的(去掉注释的内容),我就将其分别保存为water.gro,Zn.gro,CF3SO3.gro,接下来就是需要通过这三个基本的水分子结构来生成我们需要的模拟盒子,这里我就没有采用gmx的solvate指令来添加水分子,而实直接用gmx insert-molecules来添加水分子。
先插入72CF3SO3阴离子,设定盒子的大小为3.2*3.2*3.2nm:
1 gmx insert-molecules -ci CF3SO3.gro -o box.gro -nmol 72 -box 3.2 3.2 3.2
这里的-ci就表示指定输入的文件,支持gro、dpb等格式的文件,-o就表示输出的文件名,这里会生成一个box.gro文件,-nool就代表添加72个CF3SO3.gro里面的结构,-box就表示盒子的大小。接下来就是继续添加Zn离子和水分子,与这个添加CF3SO3的操作类似,但是这里还需要将我们上一个获得的box.gro文件作为盒子文件输入(也就是不再需要-box这个指令,同时保留我们已经插入的水分子):
1 2 3 gmx insert-molecules -ci Zn.gro -o box.gro -nmol 36 -f box.gro
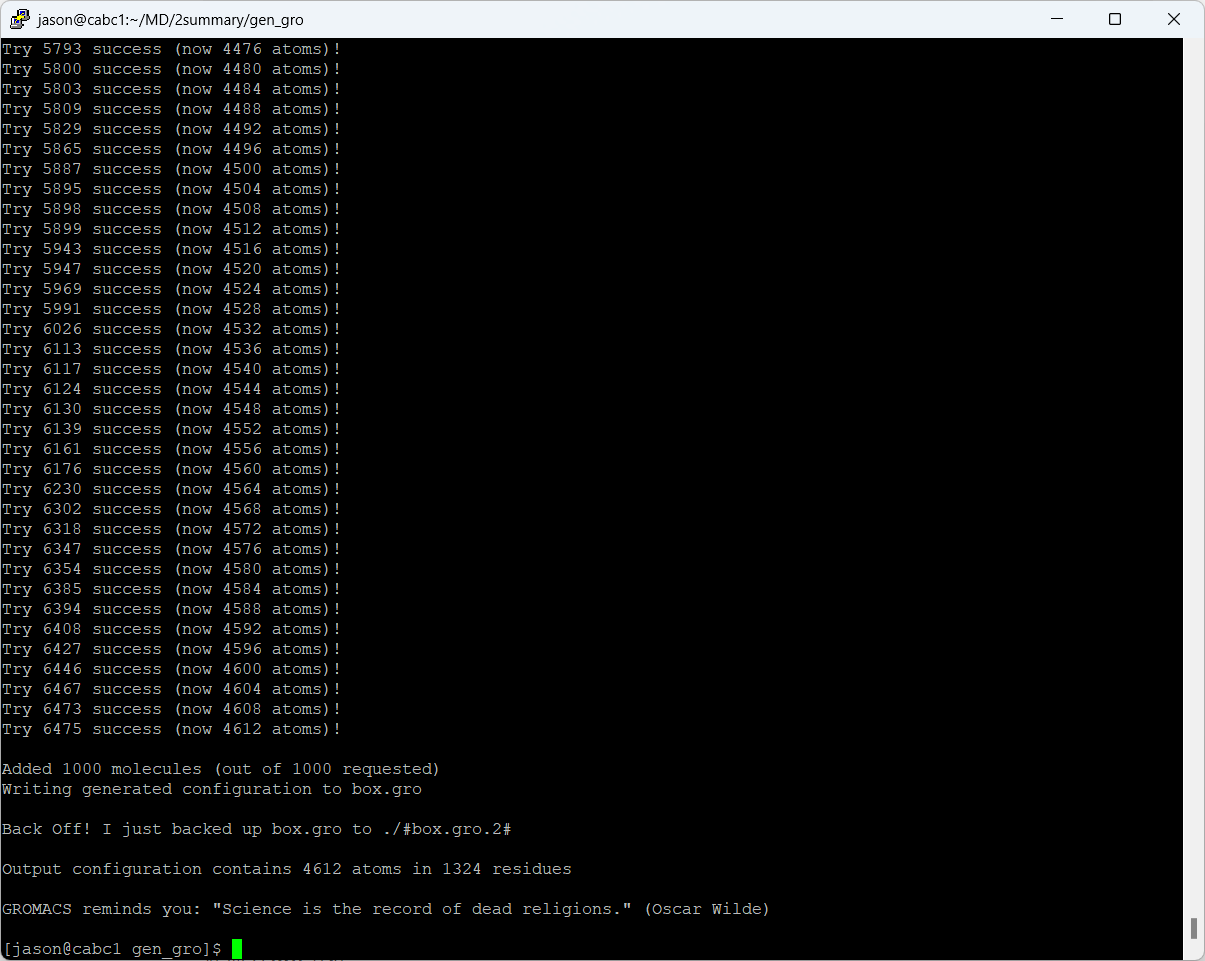
这里在插入水分子之后就代表我们所需要的盒子就已经创建好了,所以我在输出的时候将其改名为conf.gro以方便后续的进一步处理,(gromacs对gro文件的默认名称就是conf.gro,对top文件的默认名称为topl.top,所以在确定自己的gro文件和top文件完整无误之后,最好将它们的名字改为默认名,后续处理更方便)。以下是最后的界面
小结
上面这是对前期两个非常重要的文件做了一个整理归纳,这两个文件的好坏直接决定了后续计算能不能正常开展,这里写的内容就有点多了,所以我打算先分一个小结,后续的步骤在下一篇博客中讲解。