Gromacs的学习以及应用(一)
Gromacs的学习以及应用(一)
准备输入文件
盒子的分子结构文件
需要单个分子的pdb文件生成盒子的pdb文件,然后将pdb格式的盒子文件转化为gro文件。(这里看官方文档不是说pdb文件也可以作为分子结构文件?是否意味着这里无需转化)
这里我找的工作流如下:
-
通过MS建立小分子的模型结构,这里以水和甲醇两种分子为例,建立好模型后将其转化为pdb格式的文件。
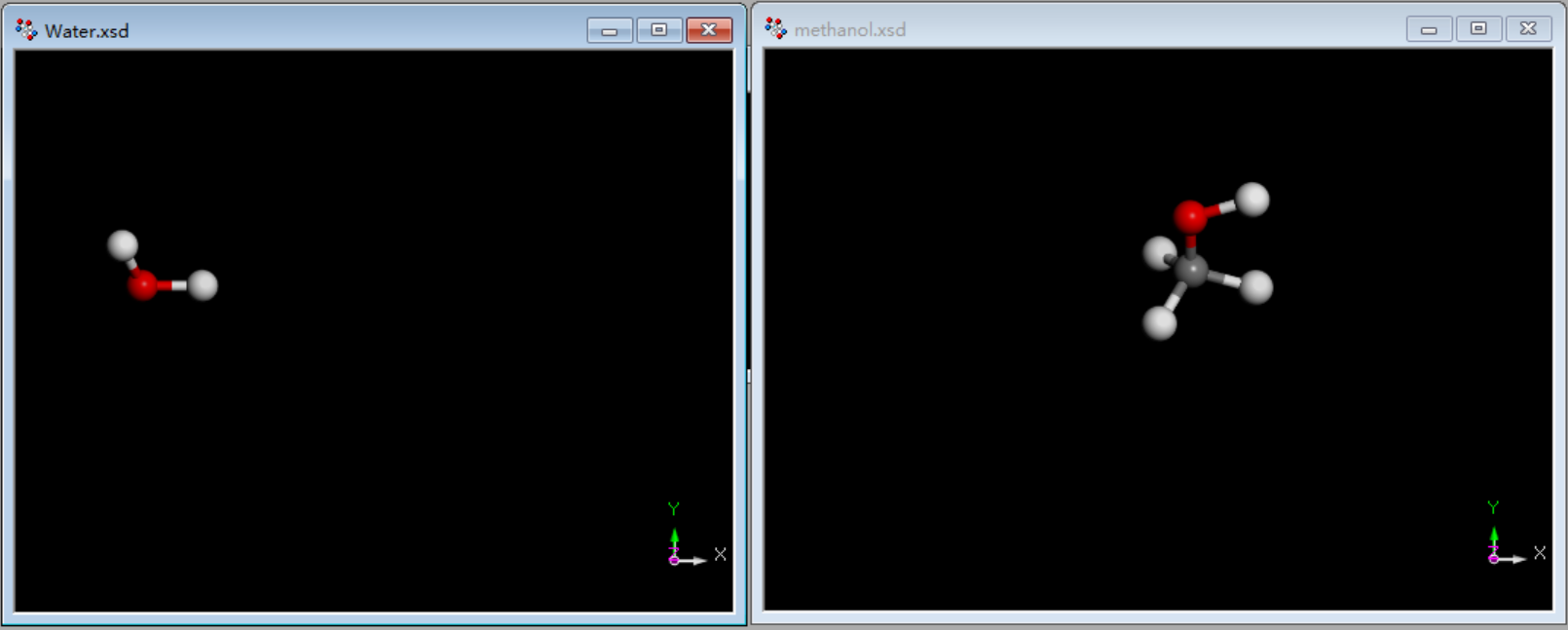
-
将文件传输到服务器上,使用提前安装好的packmol生成盒子,这里packmol的安装可以参考sobereva大佬的文章:
分子动力学初始结构构建程序Packmol的使用 - 思想家公社的门口:量子化学·分子模拟·二次元 (sobereva.com)
这里我选择甲醇和水其实也是参考了他文章中举出的例子,这里对packmol的输入文件我直接就用的是他的博客里面的输入文件,这里复制一份贴在下面
1
2
3
4
5
6
7
8
9
10
11tolerance 2.0
add_box_sides 1.2
output mix.pdb
structure water.pdb
number 1000
inside box 0. 0. 0. 40. 40. 50.
end structure
structure methanol.pdb
number 1000
inside box 0. 0. 0. 40. 40. 50.
end structure -
将获得的mix.pdb文件导入到VMD里面,就可以看到混合好的结构,这里首先是可以更改显示的模式,默认的是line模式,更改为Bond模式,然后模式是不会显示盒子的,可以输入下面的指令来显示盒子边框
1
2#显示盒子边框
pbc box
这里参考sobereva的介绍,进一步设置了一下真空层,在命令行中输入以下的指令,完成设置,然后导出数据为pdb格式
1
2
3
4
5# 重新设置box的大小,这里的单位为埃,原来的盒子大小为{40 40 50}
pbc set {40 40 110}
# 将所有原子在c周方向上移动3nm
[atomselect top all] moveby {0 0 30}
经过上面三步,就可以获得最终计算所需的pdb盒子文件,这里VMD还有许多操作是需要进一步学习的,这种命令行形式的模型处理软件应该可以实现很多便捷的操作。
接下来就是生成gro文件了,虽然据说是pdb格式的文件可以,但是我看大家都是转化成gro文件再开始计算的,我这初学者还是先照葫芦画瓢了
-
将之前VMD编辑后获得的pdb文件,命名为new_mix.pdb,传到服务器上,然后使用gromacs自带的gmx下的editconf生成gro文件,editconf的一些参数信息可以参考这篇文章
(28条消息) gmx editconf命令_weixin_43364556的博客-CSDN博客_gmx editconf
在命令行中我们输入以下代码:
1
gmx editconf -f new_mix.pdb -o new_mix.gro这里我遇到给问题,报错了,如下:
1
gmx: error while loading shared libraries: libmkl_intel_lp64.so.1: cannot open s hared object file: No such file or directory查了一下,试了不少解决方案,最后成功解决的是参考这篇文章
大概就是先find一下这个动态链接库,我是直接在根目录下的soft里面find(因为是服务器,我也不是管理员,不能去到别的用户下去find),当然,在一堆的Permission denied之下,我成功的在python的目录下找到了这个动态库,然后记下了它的目录,再按照这个目录配置了一下环境变量:
1
vim ~/.bashrc在这个文件的最后一行加上
1
export LD_LIBRARY_PATH="/soft/oneapi/intelpython/python3.7/lib:$LD_LIBRARY_PATH"注意,这里配置环境变量用的不是PATH,而是LD_LIBRARY_PATH,实测用PATH依然会报错。
然后保存文件,再输入以下指令
1
source ~/.bashrc这样就可以正常生成gro文件了
经过上面的4步这才算是生成了可以使用的分子结构文件,也就可以继续下一步了,很难想象这第一步操作竟然花了我差不多两个小时才搞定。
分子结构的拓扑文件
通过结构文件,创建top文件(拓扑文件,也可以理解为力场文件),可以直接通过gmx的pdb2gmx插件来实现,其中itp文件也会直接包含在top文件内。但是我在实际操作的时候却发现报错了,目前的原因还未知,可能是我的参数没有设定好?此外我看也有教程说pdb2gmx是适合生物大分子生成拓扑文件的,对于小分子体系并不合适,所以这里我舍弃了这个方法,以下贴出我用这个方法时输入的代码和结果:
1 | |
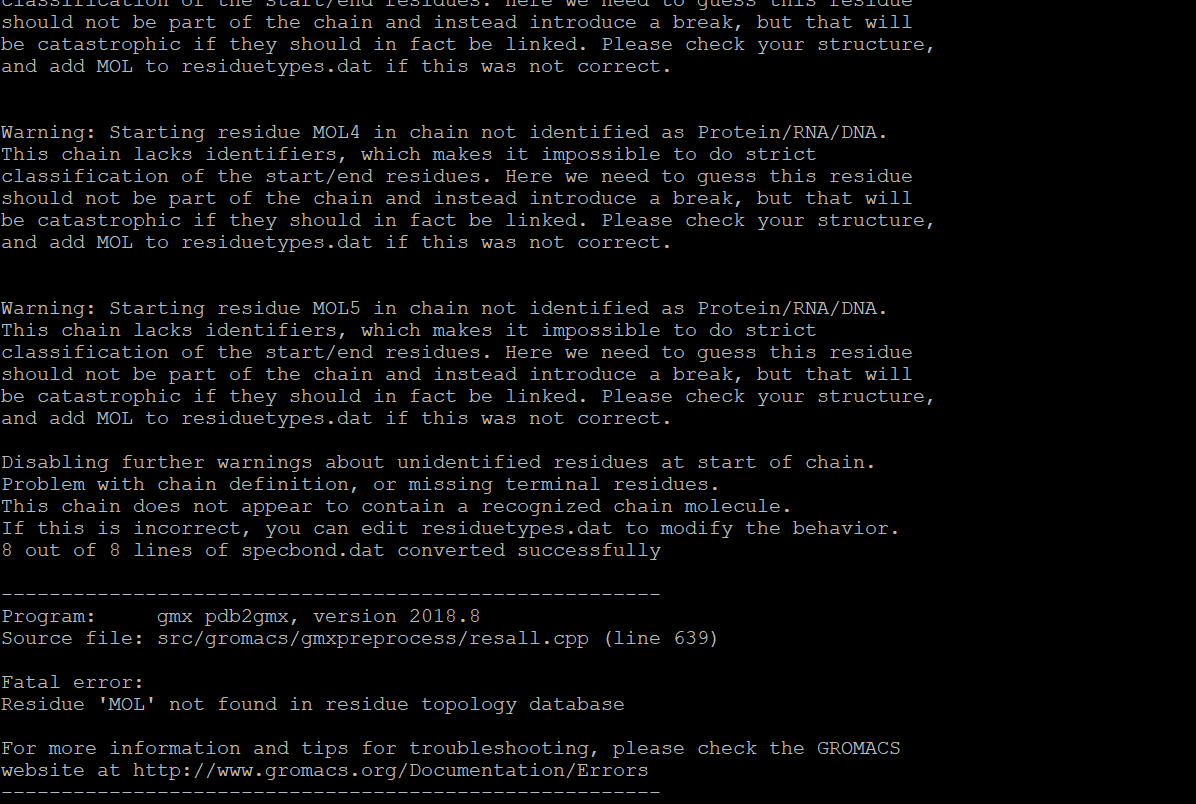
这里我遇到了很多问题,但是解决起来都相当的花费时间,而且需要深入的去学习输入文件中各项的含义,以及分子动力学的一些基本知识,所以我决定这里先暂时放在一边,搁置一些问题,采用一些可能不太准确的力场进行计算,我就用了sobtop这个小插件来生成拓扑文件,sobtop这个软件的下载以及使用可以参考这篇它的官网:
这个软件在使用上还是有些不大便利的,首先是它必须在它的这个文件目录下使用,其次每次使用的时候都需要自己手动敲一遍输入文件的路径,而且输出文件也不在输入文件的目录下,而是在这个软件所在的目录下,多少还是有点麻烦,或许后期可以自己写一个bash脚本来让它更加易用。
但参考这个软件的例子,我还是成功生成了拓扑文件,但是拓扑文件的对错我就不好说了。以后有机会了可以再好好了解一下,也可以再请教一下别人。
运行计算的参数设定文件
-
写入mdp参数文件,设定计算的细节。
这里我参考的是别人的能量最小化mdp参数,可以参考这篇帖子
求助:能量最小化的mdp文件怎么写 - 分子模拟 (Molecular Modeling) - 计算化学公社 (keinsci.com)
以下是我的mdp文件内容,这是一个凝聚态能量最小化通用的mdp文件:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19define = -DFLEXIBLE
integrator = cg
nsteps = 10000
emtol = 100.0
emstep = 0.01
;
nstxout = 100
nstlog = 50
nstenergy = 50
;
pbc = xyz
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = Cut-off
rvdw = 1.0
DispCorr = EnerPres
;
constraints = none
输入文件合并生成tpr文件
根据前面三步,我们应该就可以获得四个文件,分别为 .gro、.top、以及 .mdp文件,以及一个. top问价的附属文件 .itp( 代表自己命名的部分),这里就可以使用gromacs自带的grompp功能生成tpr文件,在命令行中输入以下的指令:
1 | |
输入这行指令后会产生两个文件,一个为mdout.mdp,这个文件就是会将所有的计算参数给罗列出来,如果你自己写的mdp文件没有规定的参数,就会用默认参数,如果你的mdp文件指定了参数,那么这里面对应的参数就会是你设置的参数。
至此,所有前期的准备就搞定了,可以开始提交任务了!
Gromac计算任务的提交
这里因为我没有提交Gromacs任务的脚本,主要是不想去麻烦老师和管理员(而且找了他们还不知道要等多少时间才能把提交任务的脚本发给我),所以我决定自己鼓捣一下这个提交任务的脚本,因为我的账号上已经有了vasp和Gaussian的脚本,所以我照着这俩脚本抄了一下,修修改改,应该也能用,注意,在提交任务的最后一行最后的em是自己生成的tpr文件的名字,比如你生成的tpr文件为mix.tpr,那么这里的em就应该改成mix:
1 | |
对了,这里参考了这篇帖子,里面提出来不少Gromacs并行计算的方法,具有一定的参考价值:
GROMACS (2019.3 GPU版) 并行效率测试及调试思路 - 分子模拟 (Molecular Modeling) - 计算化学公社 (keinsci.com)
这里想提交任务其实还是不行的,因为上面我们获取的文件Gromacs是无法直接读取的,需要根据上面的文件生成tpr文件才行,所以这里需要用到gmx grompp来生成,我在这里参考了这篇文章,里面一个人的回帖虽然没有多做描述,但是看他的脚本还是可以参考到很多有用的知识的。
求一个GROMACS提交作业的脚本,不知道怎么在服务器提交作业 - 分子模拟 (Molecular Modeling) - 计算化学公社 (keinsci.com)
这里把他给出的脚本贴出来:
1 | |
兜兜转转终于把任务给成功提交了,最后计算完成的话会再生成四个文件,其中 .edr和.trr应该是两个二进制文件,分别记录了能量和轨迹信息,还有一个 .log文件就是计算过程中输出的数据,也就是日志文件,然后还会更新一个.gro文件,就是能量最小的结构,方便进行下一步计算。
总结
以上这些内容都是在我一遍摸索的时候一遍记录下来的,前后花了我差不多7个小时,整体还是有相当多疏漏的地方,但是我的目的其实也就是想把这计算的整个流程跑通罢了,确定好整个工作流了,后面才好将它应用在实际的文章中,接下来还需要学习的东西有很多,这里我就打算通过复现文章内容的方式来进一步学习了,此外可能还得恶补一下分子动力学的内容,慢慢来吧,相较于我曾经自学vasp,现在一天就差不多摸到分子动力学软件入门的门把手了,这真的也算是一个不小的进步。