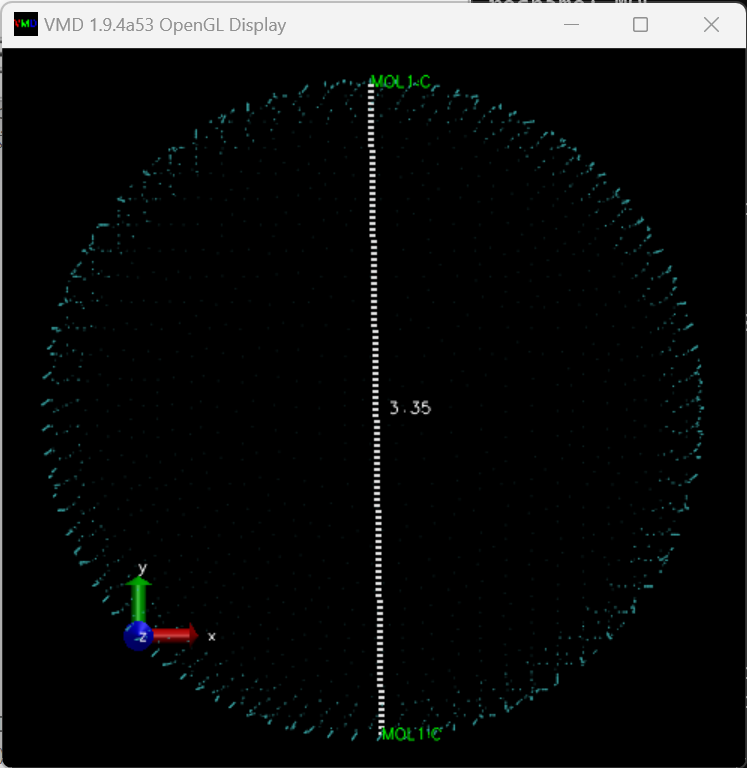
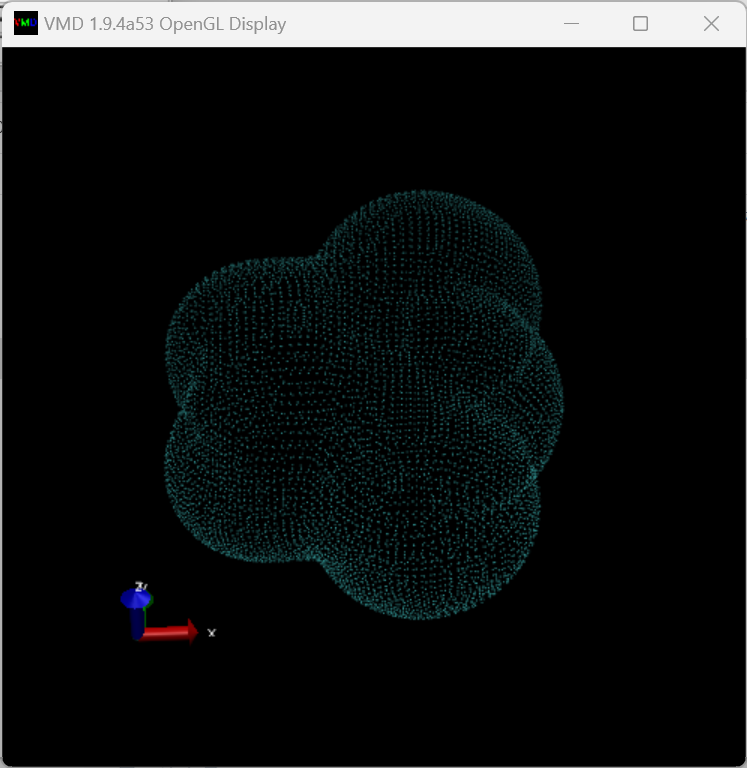
0 1 C 0.0 0.0 0.0 O 0.0 0.0 1.2584 O 0.0 0.0 -1.2584
计算与后处理
计算结果出得很快,几秒就计算完毕了, 以下是查看log文件的信息:
1 2 3 4 5 6 7 8 9 10 11 12
[jason@cabc1 new]$ tail CO2.log
I WANT TO KNOW HOW GOD CREATED THE WORLD. I AM NOT INTERESTED IN THIS OR THAT PHENOMENON, IN THE SPECTRUM OF THIS OR THAT ELEMENT. I WANT TO KNOW HIS THOUGHTS, THE REST ARE DETAILS. -- ALBERT EINSTEIN Job cpu time: 0 days 0 hours 3 minutes 46.0 seconds. Elapsed time: 0 days 0 hours 0 minutes 9.3 seconds. File lengths (MBytes): RWF= 10 Int= 0 D2E= 0 Chk= 2 Scr= 1 Normal termination of Gaussian 16 at Fri Nov 11 17:17:20 2022.
对chk文件用formchk进行转化,将其转化为fchk文件:
1 2 3 4 5 6 7 8 9
[jason@cabc1 new]$ formchk CO2.chk Read checkpoint file CO2.chk Write formatted file CO2.fchk Initial coordinates match /B/. Warning: fchk array MicOpt element 1 has value 4622945017495814144 set to -1. Warning: fchk array MicOpt element 2 has value 4625193954506324261 set to -1. Warning: fchk array MicOpt element 3 has value 4625193954506324261 set to -1. Rotating derivatives, DoTrsp=T IDiff= 1 LEDeriv= 485 LFDPrp= 0 LDFDPr= 0.
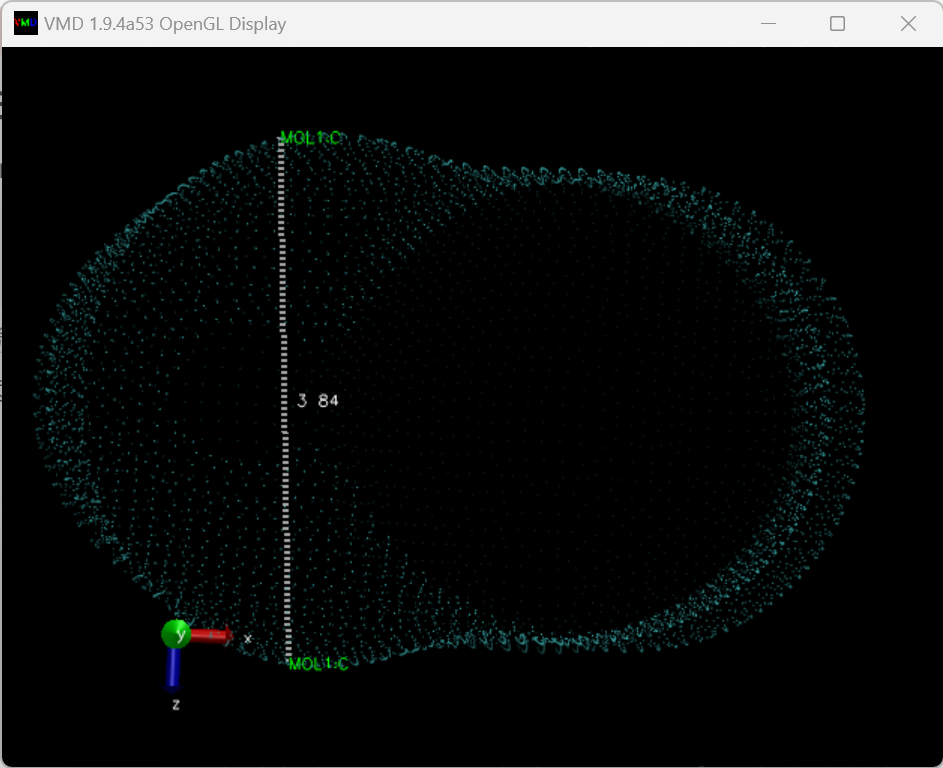
0 1 C -0.71360000 0.29750000 0.00000000 C -0.71360000 -0.52750000 0.00000000 C 0.07110000 -0.78240000 0.00000000 C 0.55600000 -0.11500000 0.00000000 O 0.07110000 0.55240000 0.00000000 O -1.38100000 0.78240000 0.00000000 O 1.38100000 -0.11500000 0.00000000 H -1.57922481 -1.15646239 0.00000000 H 0.40178325 -1.80001908 0.00000000
... A MOLECULAR SYSTEM ... (PASSES) ... FROM ONE STATE OF EQUILIBRIUM TO ANOTHER ... BY MEANS OF ALL POSSIBLE INTERMEDIATE PATHS, BUT THE PATH MOST ECONOMICAL OF ENERGY WILL BE THE MORE OFTEN TRAVELED.
-- HENRY EYRING, 1945 Job cpu time: 0 days 0 hours 8 minutes 24.3 seconds. Elapsed time: 0 days 0 hours 0 minutes 16.7 seconds. File lengths (MBytes): RWF= 32 Int= 0 D2E= 0 Chk= 4 Scr= 1 Normal termination of Gaussian 16 at Fri Nov 11 20:18:16 2022.
随后用formchk转化chk文件的格式:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
[jason@cabc1 MDD]$ formchk MA.chk Read checkpoint file MA.chk Write formatted file MA.fchk Coordinates translated and rotated. Coordinates match /B/ after translation and rotation. Warning: fchk array MicOpt element 1 has value 4622945017495814144 set to -1. Warning: fchk array MicOpt element 2 has value 4622945017495814144 set to -1. Warning: fchk array MicOpt element 3 has value 4622945017495814144 set to -1. Warning: fchk array MicOpt element 4 has value 4622945017495814144 set to -1. Warning: fchk array MicOpt element 5 has value 4625193954506324261 set to -1. Warning: fchk array MicOpt element 6 has value 4625193954506324261 set to -1. Warning: fchk array MicOpt element 7 has value 4625193954506324261 set to -1. Warning: fchk array MicOpt element 8 has value 4607217659633734768 set to -1. Warning: fchk array MicOpt element 9 has value 4607217659633734768 set to -1. Rotating derivatives, DoTrsp=T IDiff= 2 LEDeriv= 1349 LFDPrp= 0 LDFDPr= 0.